
SOMMARIO
ANALISI CHIMICA QUANTITATIVA VOLUMETRICA
*GENERAL1TA'
*
ACIDI - ALCALIMETRIA *
GENERALITA' - INDICATORI.
*TABELLA
DEGLI INDICATORI *Punto di Equivalenza
. *1) Fra acido forte e base forte
*2) Fra acido debole e base forte
*3) Fra acido forte e base debole
*4) Fra un acido forte ed una base debole bivalente
*PREPARAZIONE DELLA SOSTANZA BASE (soda)
*PREPARAZIONE DELLA SOLUZIONE DI HCI N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO (sostanza base soda)
*Il FATTORE CORRETTIVO
*PREPARAZIONE DELLA SOLUZIONE DI NaOH N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO (sostanza di riferimento HCl N/10)
*DETERMINAZIONE DEGLI ACIDI FORTI (acidità totale)
*DETERMINAZIONE DELLE BASI FORTI (alcalinità totale)
*DETERMINAZIONE DEI CARBONATI ALCALINI
*DETERMINAZIONE DEGLI IDRATI ALCALINI ACCANTO Al CARBONATI
a) Metodo di Winkler:. *
b) Metodo del doppio indicatore:
*DETERMINAZIONE DEI CARBONATI ALCALINI ACCANTO AI BICARBONATI
*DETERMINAZIONE DELL'AMMONIO (Kjeldhal)
*DETERMINAZIONE DEI FOSFATI (metodo molibdato)
*DETERMINAZIONE DEI BORATI ALCALINI
*DETERMINAZIONE DELL'ACIDO BORICO LIBERO
*DETERMINAZIONE DELL'ACIDO FOSFORICO
*DETERMINAZIONE DELLA DUREZZA TEMPORANEA DELL'ACQUA (alcalinità)
*DETERMINAZIONE DELL'ALDEIDE FORMICA
*DETERMINAZIONE DEL MERCURIO
*
ARGENTOMETRIA *
GENERALITA'
*PREPARAZIONE DELLA SOLUZIONE DI AgNO3 N/10 E DETERMINAZIONE DEL SUO FATTORE CORRETTIVO (sostanza base cloruro di sodio)
*PREPARAZIONE DELLA SOLUZIONE DI NH4CNS N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO
*DETERMINAZIONE DELLO IONE Cl- (acido cloridrico e cloruri) SECONDO MOHR
*DETERMINAZIONE DEL Cl-, Br-, I-,CN-,CNS- SECONDO VOLHARD
*DETERMINAZIONE DELLO IONE CN- (acido cianidrico e cianuri)
*
OSSIDIMETRIA
*GENERALITA'
*Le reazioni di ossidoriduzione
*Potenziale normale E°,
*PERMANGANATOMETRIA *
GENERALITA'
*PREPARAZIONE DELLA SOLUZIONE DI KMnO4 N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO (sostanza base ossalato sodico)
*DETERMINAZIONE DEL Fe++ E DEL Fe+++ SECONDO ZIMMERMANN
*DETERMINAZIONE DEL CALCIO (E DEGLI OSSALATI)
*DETERMINAZIONE DELL'ACQUA OSSIGENATA (E DEI PERCARBONATI E PERBORATI)
*DETERMINAZIONE DEL CROMO Cr METODO AL PERSOLFATO
*DETERMINAZIONE DELLO STAGNO Sn++
*IODOMETRIA *
GENERALITA'
*PREPARAZIONE DELLA SALDA D'AMIDO
*PREPARAZIONE DELLA SOLUZIONE DI TIOSOLFATO SODICO N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO (sostanza base permanganato di potassio N/10 oppure bicromato di potassio)
*DETERMINAZIONE DEL CLORO ATTIVO (ACQUA DI CLORO E IPOCLORITI)
*DETERMINAZIONE DEL RAME
*DETERMINAZIONE DELL'ACQUA OSSIGENATA
*DETERMINAZIONE DEL GLUCOSIO FEHLING
*TABELLA DI ALLIHN
*BROMATOMETRIA *
GENERALITA'
*PREPARAZIONE DELLA SOLUZIONE DI KBrO3 N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO
*DETERMINAZIONE DEL FENOLO (E ANILINA)
*DETERMINAZIONE DELL'ALLUMINIO CON 8-OSSICHINOLINA
*DETERMINAZIONE DELL'ANTIMONIO (ARSENICO E STAGNO)
*
COMPLESSOMETRIA *
GENERALITA'
*Tabella Indicatori Complessometrici
*PREPARAZIONE DELLA SOLUZIONE DI EDTA N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO (sostanza base solfato di zinco eptaidrato)
*DETERMINAZIONE DELL'ALLUMINIO
*DETERMINAZIONE DEL BARIO
*DETERMINAZIONE DEL SOLO CALCIO (in presenza di Magnesio)
*DETERMINAZIONE DEL CALCIO (Magnesio, Zinco)
*DETERMINAZIONE DEI SOLFATI
*DETERMINAZIONE DELLA DUREZZA DELLE ACQUE
*DETERMINAZIONE DEL CADMIO (ZINCO) SU UN BAGNO GALVANICO
*ANALISI CHIMICA QUANTITATIVA
L'analisi volumetrica ha, rispetto alla ponderale, il grande vantaggio della maggior rapidità. Essa si basa sulla realizzazione di reazioni complete il cui punto finale è individuabile con sufficiente precisione attraverso una variazione di colore o formazione di precipitato.
Le reazioni cosiddette complete, sono in realtà reazioni fortemente spostate in un verso. Esse sono:
1 - reazioni che comportano la formazione di un sale o composto debolmente dissociato; es: acidi-alcalimetria
H+ Cl- + Na+ OH- þ Na+ Cl- + H2O
2 Na+ CO3-- + H+ Cl- þ Na+ Cl- + Na+ HCO3-
2 - reazioni di precipitazione o sviluppo gassoso; es: argentometria
Ag+ NO3- + Na+ Cl- þ Na+ NO3- + AgCl
Na+ HCO3- + H+ Cl- þ Na+ Cl- + H2O + CO2
3 - reazioni di ossidoriduzione; es: permanganatometria
Sn++ 2 Cl- + 2 Fe+++ 6 Cl- þ Sn+++ + 4 Cl- + 2 Fe++ 4 Cl-
+7 +2 +2 +3
n. ossidazione2 KMnO4 + 10 FeSO4 + 8 H2SO4 þ K2SO4 + 2 MnSO4, + 5 Fe2(SO4)3 + 8 H2O
4 - reazioni di complessazione; es: complessometria
Ag+ NO3- + 2 Na+ 2 CN- þ Na+ NO3- + Na+ Ag(CN)2-
-NC þ Ag+ ï CN-
HNa(COOCH2)2N (CH2)2 N(CH2COO)2NaH + Ca++ 2 Cl-
þ 2 H+ Cl- + Na(COOCH2)2 N (CH2)2 N(CH2COO)2NaEDTA Ca++
I punti finali di queste reazioni, che esamineremo più da vicino a proposito delle varie parti dell'analisi volumetrica descritte più avanti, devono essere visibili attraverso una variazione di colore o formazione di precipitato oppure aggiungendo una sostanza capace di dare questi fenomeni, sostanza detta indicatore.
I reagenti fondamentali dell'analisi volumetrica sono le soluzioni a contenuto noto che devono essere preparate con grande esattezza dipendendo da quest'ultima il buon risultato delle analisi. Il loro contenuto noto è generalmente espresso come normalità cioè come equivalenti per litro.
Il peso equivalente di una sostanza è il peso molecolare diviso per la valenza e, nel caso trattisi di reazioni di ossidoriduzione, darà il peso molecolare diviso per il salto di valenza (attività protonica).
Per esempio:
HCl come acido Peso eq. = HCl /1
Na2CO3 come base = Na2CO3 /2
HNO3 come acido = HNO3 /1
+5 +2
HNO3come ossidante (NO3-/NO) = HNO3 /3
FeCl2 come sale = FeCl2 /2
+2 + 3
FeCl2 come riducente (Fe/Fe) = FeCl2 /1
+7 + 2
KMnO4 come ossidante (MnO4-/Mn++) = KMnO4 /5
Stabilito il peso equivalente, le soluzioni vengono preparate in modo da contenere una frazione nota di esso; per esempio, N/2, N/5, N/10, N/100, i ml contenenti rispettivamente 0,5, 0,2, 0, 1, 0,01 equivalenti per litro. Queste soluzioni devono essere esattamente controllate mediante una determinazione in bianco (si esegue una determinazione di cui si conosce il risultato e che si dovrà riscontrare) su una sostanza adatta detta sostanza base e, nel caso frequente in cui risultino leggermente errate, anziché correggerle materialmente si nota per ognuna di esse l'errore noto sotto forma di un fattore correttivo il cui metodo di determinazione ed uso verrà indicato caso per caso.
I metodi di analisi quantitativa volumetrica che qui di seguito saranno descritti sono ACIDI - ALCALIMETRIA - ARGENTOMETRIA, PERMANGANATOMETRIA - IODOMETRIA - BROMATOMETRIA - COMPLESSOMETRIA.
Raccolta formule:
N = n. eq. / 1l
p. eq. = pm / Val
n. eq. = m / p. eq. m = n. eq. ∙ p. eq.
N = Normalità
n. eq. = numero equivalenti
p. eq. = peso equivalente
m = massa in g
Val. = Valenza (attività protonica)
1l = 1 litro(1.000 ml)
GENERALITA' - INDICATORI - PREPARZ. DELLA SOST. BASE (SODA) - PREPARAZ. DELLA SOLUZ. DI HCI N/10 E DET. DEL FATT. CORR. (SOST. BASE SODA) - PREP. DELLA SOLUZ. DI NaOH N/10 E DET. DEL FATT. CORR. (SOSTIT. BASE HCI N/10) - DET. DEGLI ACIDI FORTI (ACIDITA' TOTALE) - DET. DELLE BASI FORTI (ALCALINITA' TOTALE) - DET. DEI CARBONATI ALCALINI - DET. DEGLI IDRATI ACCANTO Al CARBONATI (METODO DI WINKLER E DEL DOPPIO INDICATORE) - DET. DEI CARBONATI ALCALINI ACCANTO AI BICARBONATI - DET. DELL'AMMONIO (KJELDHAL) - DET. DEI FOSFATI (METODO AL MOLIBDATO) - DET. DELL'AC. BORICO LIBERO - DET. DELL'AC. FOSFORICO - DET. DELLA DUREZZA TEMPORANEA DELL'ACQUA (ALCALINITA') - DET. DELL'ALDEIDE FORMICA - DET. DEL MERCURIO.
Questo metodo di analisi volumetrica, si basa sulle reazioni di neutralizzazione fra acidi e basi usando determinate sostanze come indicatori.
E' noto che le reazioni di neutralizzazione comportano una variazione continua di pH, variazione che raggiunge la massima rapidità al punto di equivalenza, al punto cioè in cui acido e base sono presenti in equivalenza stechiometrica e si è formato il loro sale. L'indicatore avrà il compito di presentare una variazione di colore, cioè un viraggio, al punto di equivalenza.
Gli INDICATORI quindi sono sostanze coloranti capaci di assumere un diverso colore per un diverso pH. Si tratta in effetti di acidi o basi molto deboli le cui molecole hanno un colore diverso dai loro ioni: per esempio, il metilarancio è un acido debole le cui molecole presentano un colore rosa-rosso; quando esse si dissociano (in soluzione basica) generano un ione H+ ed un anione di colore giallo.
metilarancio (acido p. dimetilammino, azobenzensolfonico)
Esiste una costante di dissociazione dell'indicatore indicata con Kind la quale esprime in effetti che il rapporto fra i due colori dipende dalla conc. degli ioni H+. Per il metilarancio, sotto pH 3, prevale nettamente il rosso (molecole non dissociate), sopra pH 4,5, prevale nettamente il giallo (molecole dissociate): nel campo di pH 3-4,5, coesistono le due forme e si verifica il cosidetto viraggio. Indicando con HR questo indicatore, abbiamo:
HR Ö H+ R- [H+] [R-] / [HR] = Kind
rosso giallo
[H+] [giallo] / [rosso] = Kind [giallo] / [rosso] = Kind /[H+]
Cioè il colore dipende dalla Kind e dal pH della soluzione.
INDICATORE SOLUZIONE pH DI VIRAGGIO COLORI
Blu timolo 0,4% idroalcolica 1,2 -2,8 rosso/giallo
Violetto di metile 0,1 % acqua 0,7-3,2 giallo/viola
Tropeolina 00 0,01 % acqua 1,3-3,2 rosso/giallo
Rosso Congo 0,1 % acqua 3-5 violetto/rosso
Metilarancio 0,1 % acqua 3-4,5 rosso/giallo
Rosso di Metile 0,2% idroalcolica 4,2-6,3 rosso/giallo
Tornasole neutro estratto idroalc. 6-7 rosso/azzurro
Blu bromotimolo 0,4% idroalcolica 6-7,6 giallo/azzurro
Porpora bromocresolo 0,4% idroalcolica 5,2-6,8 giallo/porpora
Rosso alizarina 0,1 % acqua 5,5-6,5 giallo/rosa
Acido rosolico 0,1 % acqua 6,8-8 giallo/rosso
Fenolftaleina 0,5% idroalcolica 8-10 incolore/rosso
Timolftaleina 0,4% idroalcolica 10-11 incolore/azz.
Giallo alizarina GG 0, 1 % idroalcolica 10-12 giallo/lilla
Azzurro Poirrier 0,2% acqua 11-13 azz./rosso cupo
Tropeolina 0 0,5 % acqua 11- 13 giallo/verde
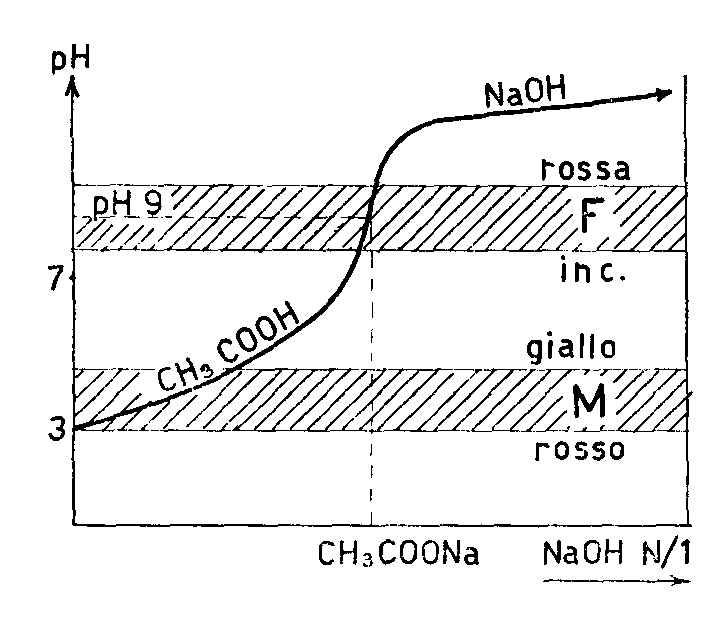
1) Fra acido forte e base forte (es.: HCl ed NaOH), il punto di equivalenza si verifica a pH 7 perchè i loro ioni sono incapaci di turbare l'equilibrio dell'acqua non presentando nessun fenomeno associativo. Il sale che formano (es.: NaCl) non dà infatti luogo ad idrolisi. Nel diagramma qui a fianco è indicato l'andamento del pH durante la neutralizzazione e le fasce di viraggio del metilarancio e della fenolftaleina.

2) Fra acido debole e base forte (es.: CH3COOH + NaOH) il punto di equivalenza si presenta ad un pH basico perché il sale che si forma (es.: CH3COONa) presenta una idrolisi con reazione basica:
H20
CH3COO- Na+ Ö CH3COOH + Na+ OH- [CH3COOH] [OH-] = Ki
[CH3COO-]
ed essendo [CH3COOH] = [OH-] perchè prodotti contemporanei della stessa idrolisi ed essendo
[CH3COO-] = c (concentrazione dell'acetato sodico in beaker al punto di equivalenza), si ottiene:
[OH-]2 / c =
Ki [OH-]
![]()
La costante di idrolisi Ki e a sua volta eguale al rapporto fra la costante dell'acqua Kw e quella dell'acido acetico Ka, come risulta facilmente dividendo membro a membro il prodotto ionico, dell'acqua per l'equilibrio dell'ac. acetico. Si ottiene in definitiva:
[OH-] = ![]()
da cui, fissato a stima il valore di c, si ricava il pH di viraggio.
Nel caso portato ad esempio del CH3COOH + NaOH, essendo Ka = 10-5 e ponendo c = 10-1 avremo:
[OH-] =
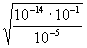 =
10-5 pH 9
=
10-5 pH 9
Nel diagramma qui a fianco indicato si vede l'andamento del pH durante la neutralizzazione e le fasce di viraggio del metilarancio (non utile) e della fenolftaleina utile in questo caso.
3) Fra acido forte e base debole (es.: HCl + NH4OH) il punto di equivalenza si presenta ad un pH acido perchè il sale che si forma (es.: NH4Cl) presenta una idrolisi con reazione acida:
H2O
NH4+ Cl- Ö NH4OH + H+ Cl- [NH4OH] [H+] = Ki
[NH4+]
ed essendo [NH4OH] = [H+] perché sono prodotti contemporanei della stessa idrolisi ed essendo [NH4+] = c (concentrazione del cloruro ammonico in beaker al punto di equivalenza) si ottiene:
[H+]2 /c = Ki
[H+] = ![]()
La costante di idrolisi Ki è a sua volta eguale al rapporto fra la costante dell'acqua Kw e quella dell'idrato ammonico Kb, come risulta facilmente dividendo membro a membro il prodotto ionico dell'acqua per l'equilibrio dell'idrato ammonico. Si ottiene in definitiva:
[H+] = ![]()
da cui, fissato a stima il valore di c; si ricava il pH di viraggio. Nel caso portato ad esempio del
HCl + NH4OH, essendo Kb = 10-5 e ponendo c = 0,1, avremo:
[H+] =![]() = 10-5
pH 5
= 10-5
pH 5
 Nel
diagramma qui sopra indicato si vede l'andamento del pH durante la
neutralizzazione e le fasce di viraggio della fenolftaleina (non utile) e del
metilarancio utile in questo caso.
Nel
diagramma qui sopra indicato si vede l'andamento del pH durante la
neutralizzazione e le fasce di viraggio della fenolftaleina (non utile) e del
metilarancio utile in questo caso.
4) Fra un acido forte ed una base debole bivalente (HCl + Na2CO3) vi sono punti di equivalenza corrispondenti riferendoci all'esempio, alla formazione di NaHCO3 e H2CO3:
H+ Cl- + 2Na+ CO3-- þ Na+ Cl- + Na+ HCO3- pH 8,3
H+ Cl- + Na+ HCO3- þ Na+ Cl- + H2CO3 pH 3,8
Nel diagramma qui a fianco indicato si vede l'andamento del pH durante la neutralizzazione e le fasce di viraggio del metilarancio e della fenolftaleina; si vede che la fenolftaleina è utile ad individuare il primo punto di equivalenza (NaHCO3 pH 8,3) mentre il metilarancio è utile per individuare il secondo (H2CO3 pH 3,8).

Nella scelta dell'INDICATORE bisogna d'altra parte ammettere che il punto di equivalenza non venga mai esattamente raggiunto calando la soluzione titolante dalla buretta, ma che tale punto venga di fatto, o superato da una ulteriore goccia o avvicinato per difetto di una goccia: tale goccia in più o in meno, modifica ovviamente il pH previsto per il sale e lo scostamento è diverso e seconda che si tratti di goccia di acido o base forti oppure di goccia di acido o base deboli e che questi siano N oppure N/10.
Operando su 50-100 ml di soluzione, tale stima, può essere approssimata nella misura di 2 unità di pH per goccia di acido o base forte N/10 (3 unità di pH per soluzioni N) ovvero nella misura di 0,5 unità di pH per goccia di acido o base debole N/10 (circa 1 unità per soluzioni N).
Esempi :
Avendo acido e base forti abbiamo:
pH del sale = 7; salto di pH in più o in meno per errore di una goccia = 2; campo utilizzabile
pH 7 ± 2 = 5 - 9. Usabili tutti gli indicatori della tabella compresi fra il Rosso Congo e la Fenolftaleina.
Avendo acido debole e base forte (CH3COOH e NaOH) abbiamo:
pH del sale = 9; salto di pH in meno per effetto dell'acido = 0,5; salto di pH in più per effetto della base = 2; campo utilizzabile pH 9 - 0,5 = pH 8,5; pH 9 + 2 = pH 11 e cioè pH 8,5 - 11. Usabile la Fenolftaleina.
Avendo acido forte e base debole (HCl e NH4OH) abbiamo:
pH del sale = 5; salto di pH in meno per effetto della goccia di acido = 2; salto di pH in più per effetto della goccia di base = 0,5; campo utilizzabile pH 5 - 2 = 3; pH 5 + 0,5 = pH 5,5; cioè pH 3-5,5. Usabili Rosso Congo, Metilarancio e Rosso di Metile ecc..
Si evita titolazione fra acido e base deboli: comunque usabili Rosso alizarina o miscela apposita di indicatori.
[H+] = ![]()


PREPARAZIONE DELLA SOSTANZA BASE (soda)
Modo di operare:
Un g circa di bicarbonato di sodio puro NaHCO3, viene riscaldato in una capsula per mezz'ora sui
250°-300° C mescolandolo col filo di platino. Quindi si conserva in essiccatore il Na2CO3 formatosi.
PREPARAZIONE DELLA SOLUZIONE DI HCI N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO (sostanza base soda)
Il peso equivalente dell'HCl è 36,465; una sua soluzione N/10 dovrà perciò contenere g 3,6465 di HCl per litro di soluzione.
Ammettiamo di doverne preparare 1 litro (1000 ml) e di disporre in laboratorio di HCl conc.
d = 1,18 concentrazione 36,23% :
g 3,6465 di HCl (100%) corrispondono a g 3,6465 × 100 / 36,23 = g 10,06 di HCl al 36,23%
g 10,06 di HCI al 36,23% d = 1,18 corrispondono a ml (10,06 : 1,18) = ml 8,5 di HCl conc. da prelevare
Modo di operare:
Prelevare perciò con una buretta o con una pipetta munita di gomma aspirante, ml 8,5 di HCl conc. d = 1,18 e calarli in un matraccio tarato da 1000 ml e portare a volume con acqua distillata.
E' assai improbabile che la soluzione cloridrica così preparata sia esattamente N/10; essa esige in ogni modo un controllo che viene eseguito mediante una prova in bianco, cioè facendo agire la soluzione presunta N/10 su un campione di sostanza base (soda) esattamente pesata:
- se la soluzione preparata sarà esattamente N/10, si dovranno trovare, per titolazione, i grammi di soda pesati;
- se la soluzione preparata, non sarà esattamente N/10 caso molto comune, si avrà dalla titolazione un risultato diverso dai grammi di soda pesati: tale diverso risultato ci permetterà di conoscere l'entità dell'errore contenuto nella soluzione preparata di HCl.
Si procede comunque nel seguente modo:
Modo di operare:
Si pesa esattamente una quantità di soda Na2CO3 purissima compresa fra g 0,1 e 0,2.
Ammettiamo di averne pesati g 0,1865.
Si scioglie la soda pesata in 50 ml circa di acqua distillata e si aggiungono 3 gocce di metilarancio (colorazione gialla): si titola con HCl N/10 da controllare fino a viraggio debolmente rosso. I ml di HCl N/10 in esame usati in tale operazione vengono denominati ml pratici.
Ora, essendo il peso equivalente della soda eguale a (PM Na2CO3 : 2)=53,002, e tenendo presente che si opera con una soluzione N/10 (53,002 : 10) = 5,3002, per i g 0,1865 di soda pesati e titolati, avremmo dovuto usare i ml risultanti dalla seguente proporzione:
1.000 : 5,3002 = X : 0, 1865
Gli x ml vengono denominati ml teorici.
Il rapporto ml teorici / ml pratici = fattore correttivo
La soluzione cloridrica da noi preparata, avrà quindi la seguente denominazione:HCl N/10 fattore correttivo . . .
Ogni qualvolta si userà questa soluzione, bisognerà moltiplicarne i ml usati per tale fattore
Se il f.c. è eguale ad uno (ml teorici = ml pratici), significa che la soluzione è esatta ed ovviamente viene usata senza correzione.
Se il f.c. è minore di uno (ml teorici minori dei ml pratici), significa che la soluzione preparata è più diluita del N/10. Quindi, nelle analisi, se ne userà un maggior numero di ml e tale maggior numero viene riportato al vero appunto moltiplicandolo per il f.c.
Se il f.c. è maggiore di uno (ml teorici maggiori dei ml pratici), significa che la soluzione preparata è più concentrata del N/10. Quindi nelle analisi, se ne userà un minor numero di ml e tale minor numero viene riportato al vero appunto moltiplicandolo per il f.c.
PREPARAZIONE DELLA SOLUZIONE DI NaOH N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO (sostanza di riferimento HCl N/10)
Il peso equivalente della NaOH è 40,005; una sua soluzione N/10 dovrà perciò contenere g 4,0005 per litro (1000 ml) di soluzione.
Modo di operare:
Pesare, quindi, dentro un pesafiltri tarato g 4,1 - 4,2 di NaOH solida e pura e porli in un matraccio tarato da 1000 ml : portare a volume con acqua distillata, passando più volte l'acqua distillata attraverso il pesafiltri.
La soluzione così preparata, sarà approssimativamente N/10. La NaOH usata è leggermente superiore al peso equivalente, poichè essa contiene assai spesso impurezze per assorbimento di CO2, e H2O.
Comunque, tale soluzione esige la determinazione del suo fattore correttivo, che si esegue nel seguente modo:
Modo di operare:
In un beaker, si pongono 20 ml di HCl N/10 f.c. noto esattamente misurati con una pipetta, si diluisce con acqua distillata fino a circa 50 ml e vi si aggiungono 5 gocce di fenolftaleina quale indicatore: la soluzione rimarrà incolore. Tale soluzione cloridrica, viene titolata con la NaOH N/10 in esame calata da una buretta, fino a colorazione leggermente rossa persistente un minuto.
I ml di NaOH N/10 in esame usati in tale operazione, vengono denominati ml pratici.
Invece, 20 ml, di HCl N/10 moltiplicati per il loro fattore correttivo noto vengono denominati ml teorici.
Il rapporto ml teorici / ml pratici = fattore correttivo del NaOH.
La soluzione sodica da noi preparata, avrà quindi la seguente denominazione:
NaOH N/10 fattore correttivo ...
Ogni qualvolta si userà questa soluzione, bisognerà moltiplicarne i ml usati per tale fattore.
L'operazione, può essere ripetuta più volte assumendo come valore definitivo la media dei valori ottenuti.
Sarà bene ricordare, che la colorazione rossa della fenolftaleina ottenuta al viraggio, generalmente scompare nel giro di qualche minuto: ciò è dovuto all'assorbimento di CO2 dall'aria ambiente e conseguente formazione nella soluzione di acido carbonico H2CO3 il quale, pur essendo un acido debole, è
sufficiente ad inacidire la soluzione in modo sensibile alla fenolftaleina, la quale pertanto si decolora.DETERMINAZIONE DEGLI ACIDI FORTI (acidità totale)
Il campione di acido forte, viene prelevato a peso, pesandolo dentro un pesafiltri precedentemente tarato, oppure, se è nota esattamente la densità, anche a volume con una pipetta munita di pera di gomma per l'aspirazione. La quantità di campione da prelevare, deve risultare da un calcolo preventivo, allo scopo di poter usare, nella titolazione, 20 - 40 ml di soluzione titolata.
Reagenti necessari:
NaOH N/10 f.c. noto Fenolftaleina
Modo di operare:
Si calcolano preventivamente i g di prodotto necessari per preparare un litro di soluzione approssimativamente N/10, (se si usano soluzioni titolanti N/10) conoscendo la concentrazione approssimata dell'acido in esame, eventualmente dedotta con un densimetro.
Per esempio sia da esaminare un H2SO4 di concentrazione approssimata 90%.
Per preparare 1 litro di soluzione N/10 sono necessari g 4,9 di H2SO4 assoluto e cioè :
90 : 100 = 4,9 : x; x = 5,44 g di H2SO4 in esame.
Tenendo conto di tale indicazione si preleva il campione su pesafiltri tarato.
Sia la pesata (in pesafiltri) di g 5,857.
Tale quantità viene posta in un matraccio tarato da 1000 ml e portata a volume con acqua distillata: si prelevano 25 ml di tale soluzione diluita e si titolano con NaOH N/10 f.c. noto fino a viraggio della fenolftaleina: ammettiamo di usare 26 ml di NaOH N/10.
Si moltiplicano per il f.c. e quindi si calcola l' H2SO4 assoluto.
1.000 : 4,9 = (26 . f.c.) : x
x = g di H2SO4 assoluto = g 0,1274 (se f.c. = 1)
Questi g 0,1274 vengono moltiplicati per 40 perchè i 25 ml su cui si è operato rappresentano la quarantesima parte (25/1000) dell'intera soluzione: avremo così g 0,1274 × 40 = g 5,096 e perciò la concentrazione dell'acido in esame sarà:
5,857 : 5,096 = 100 : x x = 87%
P. eq. HCl = (PM HCl) = 36,465
P. eq. HNO3 = (PM HNO3) = 63,016
P. eq. H2SO4 = (PM H2SO4 : 2) = 49,040
DETERMINAZIONE DELLE BASI FORTI (alcalinità totale)
Le basi forti (soda e potassa caustica, calce, magnesia), sono spessissimo impure per carbonati e umidità e, tali impurezze, non sono sempre uniformemente distribuite: conviene dunque prelevare campioni superiori al grammo affinchè il campione rappresenti la composizione della base esaminata. D'altra parte, nella titolazione, bisogna tenere conto della capacità della buretta (50 ml) e della normalità dell'HCl che impiegheremo (N/10).
Reagenti necessari:
HCl N/10 f.c. noto Metilarancio
Modo di operare:
Pesare perciò esattamente in un pesafiltri, una quantità prossima alla decima parte del peso equivalente della base in esame, scioglierli in un matraccio tarato da 1000 ml, portare esattamente a volume con acqua distillata ed operare su 25 ml di tale soluzione titolandoli con HCl N/10 f.c. noto al metilarancio.
I g di base assoluta, risultano dalla proporzione seguente:
1.000 : p. eq. base/10 = ml usati e corretti : X
Questi X g moltiplicati per 40, costituiscono il peso di base presente sul campione pesato.
P. eq. NaOH = (PM NaOH) = 40,005
P. eq. KOH = (PM KOH) = 56,104
DETERMINAZIONE DEI CARBONATI ALCALINI
I carbonati alcalini, sono solubili e titolabili direttamente con HCl; essi, in definitiva, si comportano come una base forte poichè l'acido carbonico è debole e volatile:
X2C03 + 2 HCl þ 2 XCl + H2O + CO2
La loro titolazione è perciò assai semplice:
Reagenti necessari:
HCl N/10 f.c. noto Metilarancio
Modo di operare:
Si pesa esattamente una quantità prossima a g 0,2 se si tratta di soda o g 0,3 se si tratta di potassa (oppure quantità dieci volte superiori che verranno portate in un matraccio da 250 ml, portate a volume con acqua distillata, operando poi su 25 ml di tale soluzione).
Comunque, il campione viene posto in un beaker in un volume di circa 50 ml e titolato al metilarancio con HCl N/10 f.c. noto.
P. eq. Na2CO3 = (PM Na2CO3 : 2) = 53,002
P. eq. K2CO3 = (PM K2CO3 : 2) = 69, 101
DETERMINAZIONE DEGLI IDRATI ALCALINI ACCANTO Al CARBONATI
Metodo Winkler e del doppio indicatore
Gli idrati alcalini (soda e potassa caustiche NaOH e KOH), sono sempre impuri di CO2 e H2O che assorbono dall’aria; essi, perciò contengono sempre una certa quantità di carbonato che si forma nella reazione:
2 NaOH + CO2 þ Na2CO3 + H2O
Interessa perciò determinare il carbonato accanto all'idrato e ciò viene eseguito sia col metodo di Winkler che col metodo del doppio indicatore.
a) Metodo di Winkler: consiste nel determinare l'alcalinità totale mediante una titolazione al metilarancio con HCl N/10 f.c. noto, ottenendosi quindi l’idrato + il carbonato.
Quindi, su un altro campione, si determina il solo idrato, aggiungendo un eccesso di BaCl2, il quale converte tutto il carbonato alcalino in carbonato di bario insolubile e perciò non titolabile
(Na2CO3 + BaCl2 þ 2 NaCl + BaCO3), mentre l'idrato alcalino, si trasforma in una equivalente quantità di idrato di bario che è titolabile
(2 NaOH + BaCl2 þ 2 NaCl + Ba(OH)2).
La differenza fra le due titolazioni rappresenterà ovviamente il carbonato.
Reagenti necessari:
HCl N/10 f.c. noto BaCl2 . 2H2O solido
Metilarancio Fenolftaleina
Modo di operare:
Si pesa esattamente dentro un pesafiltri, precedentemente tarato, una quantità di prodotto prossima ad un grammo se si tratta di NaOH, o a g 1,4 se si tratta di KOH, si porta in un matraccio tarato da 250 ml portando esattamente a volume con acqua distillata.
Con una pipetta, si prelevano esattamente 50 ml di tale soluzione, si trasportano in un beaker e si titolano al metilarancio con HCl N/10 f.c. noto.
I ml usati, corretti col f.c. e moltiplicati per 5, rappresentano tutto l’idrato e tutto il carbonato.
Si prelevano con una pipetta, altri 50 ml di soluzione preparata si portano in un beaker, si diluiscono con circa 100 ml acqua distillata (diluizione necessaria per evitare la precipitazione del Ba(OH)2 che è poco solubile) e si aggiungono 3-4 g di BaCl2 · 2H2O. Si rimescola fino a soluzione completa del cloruro di bario e precipitazione completa del carbonato di bario. Si lascia a se 10' per far depositare il carbonato di bario e quindi si titola fino a decolorazione di fenolftaleina con HCl N/10 f.c. noto.
I ml usati, corretti col f.c. e moltiplicati per 5, rappresentano il solo idrato.
Ovviamente, la differenza fra le due titolazioni, rappresenterà il solo carbonato.
P. eq. NaOH = 40,005 P. eq. Na2CO3 = 53,002
P. eq. KOH = 56,104 P. eq. K2CO3 = 69, 101
b) Metodo del doppio indicatore: si tratta di eseguire una unica titolazione su un unico campione con HCl titolato di fattore noto usando come indicatori sia il metilarancio che la fenolftaleina allo scopo di poter cogliere due reazioni successive che interessano il carbonato alcalino.
Infatti, mentre l'idrato alcalino viene regolarmente salificato dall'HCl calato dalla buretta, il carbonato alcalino, subisce l'azione dell'HCl in due fasi successive e distinguibili per mezzo degli indicatori.
Le due reazioni sono:
Na2CO3 + HCl þ NaCl + NaHCO3 bicarbonato di sodio
NaHCO3 + HCl þ NaCl + H2O + CO2
Ora, il Na2CO3 (e naturalmente più ancora il NaOH), che di per sé presenta un pH 10 reagisce basico sia alla fenolftaleina (rossa) che vira a pH 8-10, meglio ancora, al metilarancio che vira a pH 4-5.
Il bicarbonato di sodio, invece risulta acido rispetto alla fenolftaleina (che diventa incolore), ma presentando di per se un pH 8, è ancora basico rispetto al metilarancio (che rimane giallo). Solo, proseguendo l'aggiunta di HCl fino a completa distruzione del bicarbonato, virerà anche il metilarancio che diviene rosso.
Reagenti necessari:
HCl N/10 f.c. noto Fenolftaleina Metilarancio
Modo di operare:
Un campione di prodotto esattamente pesato in un pesafiltri (intorno ad 1 grammo se si tratta di soda caustica e a g 1,4 se si tratta di potassa caustica), viene introdotto in un matraccio tarato da 250 ml e si porta a volume con acqua distillata esattamente.
Si prelevano 50 ml di tale soluzione con una pipetta, portandoli in un beaker; si aggiungono 3 gocce di fenolftaleina e si titola con HCl N/10 f.c. o fino a decolorazione
I ml usati e corretti, rappresentano, tutto l’idrato e metà del carbonato.
Alla stessa soluzione, si aggiungono 3 gocce di metilarancio e si prosegue la titolazione con lo stesso
HCl N/10 f.c. noto fino a viraggio lievemente rosso.
I ml usati e corretti, rappresentano l’altra metà del carbonato.
In conclusione, il doppio dei secondi ml rappresentano il carbonato, mentre i primi ml meno i secondi rappresentano l'idrato. I valori ottenuti, vanno naturalmente moltiplicati per 5.
P. eq. NaOH = 40,005 P. eq. Na2CO3 = 53,002
P. eq. KOH = 56,104 P. eq. K2CO3 = 69, 101
DETERMINAZIONE DEI CARBONATI ALCALINI ACCANTO AI BICARBONATI
Anche questa determinazione, come la precedente, si basa sull'uso dei due indicatori: la fenolftaleina che vira sul pH 9 ed il metilarancio, che vira sul pH 4. Infatti, i carbonati alcalini presentano un pH circa 10 e sono basici di fronte ai due indicatori, mentre i bicarbonati alcalini, presentano un pH circa 8 e sono acidi rispetto alla fenolftaleina, ma ancora basici di fronte al metilarancio.
Ora essendo che la soda commerciale Na2CO3 è normalmente impura per bicarbonato di sodio NaHCO3, è possibile determinare il titolo di tale soda commerciale mediante una titolazione con HCl di nota normalità usando i due indicatori.
Chimicamente avremo:
Na2CO3 + HCl þ NaCl + NaHCO3 a questa reazione partecipa metà del carbonato
e finisce quando vira la fenolftaleina.
NaHCO3 + HCl þ NaCl + H2O + CO2 a questa reazione partecipa l’altra metà del
carbonato e il bicarbonato impurezza finisce con il viraggio del metilarancio.
Reagenti necessari:
HCl N/10 Fenolftaleina Metilarancio
Modo di operare:
Un campione di soda commerciale sull'ordine di g 0,2 viene esattamente pesato in vetrino, portato in un beaker e sciolto in circa 50 ml di acqua distillata. Si aggiungono 5 gocce di fenolftaleina (soluzione rossa) e si titola con HCl N/10 f.c. noto fino a decolorazione.
I ml usati e corretti, rappresentano metà del carbonato.
Alla soluzione, si aggiungono 3 gocce di metilarancio e si prosegue la titolazione fino a viraggio lievemente rosso.
I ml usati e corretti rappresentano l'altra metà del carbonato più il bicarbonato.
P. eq. Na2CO3 = 53,002 P. eq. NaHCO3 = 84,015
P. eq. K2CO3 = 69, 101 P. eq. KHCO3 = 100, 114
DETERMINAZIONE DELL'AMMONIO
(Kjeldhal)I sali ammonici sono decomponibili con idrato sodico in ammoniaca ed acqua.
NH4Cl + NaOH þ NaCl + H2O + NH3
L'ammoniaca NH3, distilla per lieve riscaldamento e può essere raccolta in una misurata soluzione di acido di normalità nota, in modo tale da salificarlo parzialmente (2NH3 + H2SO4 þ (NH4)2SO4).
Titolando con NaOH di normalità nota l'acidità residua della soluzione di raccolta, si può risalire per differenza all'ammoniaca NH3 distillata dal sale ammonico esaminato.
Si opera come segue:
Reagenti necessari:
HCl N/10 f.c. noto NaOH N/10 f.c. noto
Metilarancio NaOH al 10%
Modo di operare:
Si pesa esattamente un campione di sale ammonico prossimo a g 0,2 se si tratta di cloruro ammonico ed a g 0,5 se si tratta di solfato ammonico (bisogna che l'NH3, che indicativamente si svilupperà, non superi l'equivalente di 50 ml di soluzione N/10).
Il campione pesato esattamente, viene introdotto nel distillatore di Kjeldahl e nell'imbuto a rubinetto sovrastante, si pongono 100 ml circa di NaOH al 10%. Il tubo di sviluppo dei vapori deve pescare in 50 ml esatti di HCl (o H2SO4) N/10 f.c. noto.
Si introducono i ml di NaOH 10% sul sale ammonico e si riscalda a debole fiamma iniziale, quindi a piccola ebollizione fino a ridurre ad un terzo la massa da distillare.
Precauzioni utili:
- Mettere delle palline di vetro del distillatore per evitare sussulti.
- Aggiungere altri 100 ml circa di NaOH 10% per aumentare il volume da distillare.
- Mettere nella beuta contenente i 50 ml di acido N/10 f.c. noto 5 gocce di metilarancio che, rimanendo
rosso, ci assicura che l'NH3, distillata non superi l'acido che la riceve.
Quindi si titola al metilarancio la soluzione di raccolta, usando NaOH N/10 f.c. noto. I 50 ml usati di acido N/10 corretti col f.c. meno i ml usati e corretti di NaOH N/ 10 di titolazione, ci danno i ml Corrispondenti all'ammonio.
 P. eq. NH4+
= 18,04
P. eq. NH4+
= 18,04
P. eq. NH3 = 17,032 P. eq. N = 14,008 P. eq. NH4Cl = 53,496
DETERMINAZIONE DEI FOSFATI (metodo molibdato)
La determinazione dei fosfati, particolarmente significativa per stabilire il valore dei fertilizzanti fosfatici, può essere eseguita con buoni risultati precipitando l'anione fosforico con molibdato ammonico (NH4)2MoO4 in ambiente notevolmente nitrico; il precipitato che si forma è il fosfomolibdato ammonico, giallo cristallino:
(NH4)3PO4 . 12 MoO3 . 2HNO3 . H2O
che filtrato e lavato per liberarlo dell'acido nitrico, viene poi sciolto con un eccesso noto di NaOH N/10 f.c. noto. Gli equivalenti di base necessari al disciogliersi di una mole di precipitato sono: 24 per le 12 MoO3 e 2 per il PO4 che va in soluzione come HPO4-- (e non come PO4--- ): totale 26; togliendo 3 equivalenti di base già presenti ( i tre ammoni), restano 23 equivalenti di NaOH necessari alla dissoluzione stechiometrica di una mole di precipitato. Ciò significa che l'equivalente del fosforo P sarà P/23:
(NH4)3PO4 . 12 MoO + 23 NaOH þ 3 NH4+ + 23 Na+ + HPO4-- + 12 MoO4-- + 11 H2O
Si titola infine l'eccesso di NaOH con HCI N/10 f.c. noto alla fenolftaleina al cui viraggio (pH 9-10) vi è la certezza che il P è in soluzione come anione bivalente HPO4--.
N.B. Dato il piccolo valore dell'equivalente PO4/23, P/23, P2O5/46, la soluzione in esame o comunque l'aliquota su cui si opera dovrà contenere intorno a g 0,004 di P oppure usare soluzioni N/1.
Reagenti necessari
:HCl N/10 f.c. noto NaOH N/10 f.c. noto HNO3 conc.
(NH4)2MoO4, al 7 % in HNO3 1:7 NH4NO3 tornasole azzurro
Modo di operare:
La soluzione in esame contenente i fosfati (intorno a g 0,004 di P oppure g 0,04 usando sol. N/1) viene
acidificata con 20 ml di HNO3, conc. ed aggiunta di 40 ml di molibdato ammonico (al 7% in HNO3 al 10%) e di 10-20 ml di nitrato ammonico NH4NO3 in soluzione concentrata. Si fa bollire fino a precipitazione e si lascia poi a sè a freddo per una mezz'ora. Si decanta su filtro a fascia bianca il liquido sovrastante e si lava il precipitato, prima per decantazione e poi sul filtro con acqua distillata contenente l’1% di nitrato ammonico fino a scomparsa dalle acque di lavaggio della reazione acida (usare tornasole azzurro). Non vale la pena di preoccuparsi eccessivamente dei residui di precipitato rimasti attaccati alle pareti del beaker perchè in questo stesso beaker si introduce il filtro col precipitato trattandolo con 50 ml esatti di NaOH N/10 f.c. noto e scaldando fino a completa dissoluzione del precipitato. Si titola alla fenolftaleina con HCl N/10 f.c. noto.
(50 ml · f.c. NaOH) - (ml di titolazione · f.c. HCl) = ml corrispondenti ai fosfati
1.000 : P. eq./10 = ml corrispondenti : X
P. eq. P = P/23 = 1,347 P. eq. PO4 = PO4 /23 = 4,13 P. eq. P2O5 = P2O5 /46 = 3,086
DETERMINAZIONE DEI BORATI ALCALINI
L'acido borico H3BO3, è un acido tanto debole da non avere la capacità di arrossare il metilarancio. Approfittando della assoluta indipendenza del metilarancio dall'acido borico, è possibile titolare le basi alcaline dei borati alcalini come se fossero sole.
Si opera nel seguente modo.
Reagenti necessari:
HCl N/10 f.c. noto Metilarancio
Modo di operare:
Un campione esattamente pesato di borato alcalino (prossimo a g 0,5 se si tratta di borace
Na2B4O7 . 10 H2O), viene portato in un beaker con 50-100 ml di acqua distillata.
Si titola al metilarancio con HCl N/10 f.c. noto.
P. eq. Na2B4O7 . 10 H2O = (Na2B4O7 . 10 H2O : 2) = 190,72
DETERMINAZIONE DELL'ACIDO BORICO LIBERO
L'acido borico, di per sè non è titolabile perchè data la sua debolezza, non ha azione sugli indicatori. Invece, in presenza di glicerina o mannite o sostanze pluriossidrilate in genere, esso diventa titolabile perché forma degli acidi monovalenti complessi del tipo

sufficientemente forti da essere titolati alla fenolftaleina con NaOH di normalità nota.
Si opera nel seguente modo:
Reagenti necessari
:NaOH N/10 f.c. noto Mannite Fenolftaleina
Modo di operare:
Si pesa esattamente un campione di acido borico H3BO3 in esame prossimo a g 0,2 lo si porta in un beaker con 100 ml circa di acqua distillata e vi si aggiunge un grammo circa di mannite pura. Su tale soluzione, si esegue una titolazione con NaOH N/10 f.c. noto alla fenolftaleina.
(A viraggio rosso ottenuto, si aggiunge ancora un po' di mannite; se la colorazione rossa dovesse scomparire, bisogna continuare la titolazione con NaOH N/10 f.c. noto fino a nuovo viraggio. Quindi controllare nuovamente i ml).
P. eq. H3BO3 = (PM H3BO3) = 61,84
(Il P. eq. è eguale al PM perchè ogni molecola di H3BO3 forma con la mannite una molecola di acido complesso
monobasico).DETERMINAZIONE DELL'ACIDO FOSFORICO
L'acido fosforico H3PO4 e un acido tribasico che per aggiunta progressiva di NaOH subisce dissociazione ionica graduale:
H3PO4 « H+ + H2PO4- (K1 = 7,5 . 10-3 pH da NaH2PO4 = 4,5)
H2PO4- « H+ + HPO4-- (K2 = 6,3 . 10-8 pH da Na2HPO4 = 9,5)
HPO4-- « H+ + PO4--- (K3 = 4 . 10-13 pH da Na3PO4 = 11,5)
Perciò, si può titolare l'acido fosforico come acido monobasico (I equilibrio) mediante un indicatore che viri intorno a pH 4,5 come il metilarancio, oppure titolarlo come acido bibasico (II equilibrio) mediante un indicatore che viri intorno a pH 9,5 come la fenolftaleina.
L'acido fosforico, come tribasico (III equilibrio), è difficilmente titolabile, perché il pH di viraggio è vicino a quello della NaOH di titolazione.
Si opera nel seguente modo:
Reagenti necessari:
NaOH N/10 f.c. noto Metilarancio
Fenolftaleina KH2PO4 NaCl
Modo di operare:
Un campione, precedentemente calcolato in forma indicativa in modo da formare 250 ml di soluzione circa M/10 (g 2,5 circa di acido assoluto), viene portato in un matraccio da 250 ml e portato a volume con acqua distillata.
Su 20 ml di tale soluzione, portata in un beaker ed aggiunta di 3 gocce di metilarancio si esegue una titolazione con NaOH N/10 f.c. noto fino a viraggio leggermente giallo. Meglio, per individuare il colore di viraggio, usare una soluzione di confronto di KH2PO4 circa 0,1 N contenente 3 gocce di indicatore metilarancio.
I ml usati e corretti, rappresentano l’H3PO4 considerato monobasico.
P. eq. H3PO4 = (PM H3PO4) = 98
Alla stessa soluzione virata al giallo, si aggiungono 5 gocce di fenolftaleina che rimarrà incolore e 5 g di cloruro sodico NaCl: quindi si prosegue la titolazione fino al rosa con la stessa NaOH f.c. noto.
I ml usati e corretti, rappresentando il secondo H’ fosforico e, la totalità dei ml usati e corretti di NaOH, rappresentano l' H3PO4 considerato bibasico.
P eq. H3PO4 = (PM H3PO4 : 2) = 49
DETERMINAZIONE DELLA DUREZZA TEMPORANEA DELL'ACQUA (alcalinità)
La durezza di un'acqua, è proporzionata al contenuto in sali: in genere, un'acqua è considerata dura se contiene più di mezzo grammo di sali per litro oppure dolce, se ne contiene meno.
I sali più comuni che costituiscono durezza, sono i bicarbonati, i cloruri ed i solfati di calcio e magnesio.
All'ebollizione, i bicarbonati, si trasformano in carbonati insolubili
(Ca(HCO3)2 þ CaCO3, + H2O + CO2)
e cessano di costituire durezza: perciò, i bicarbonati costituiscono la durezza temporanea (scompare all'ebollizione) mentre i cloruri ed i solfati, che all'ebollizione rimangono in soluzione, costituiscono la durezza permanente. La somma delle due durezze, costituisce la durezza totale.
La determinazione della durezza temporanea, cioè dei bicarbonati alcalino (che sono solubili), viene eseguita mediante semplice titolazione al metilarancio con HCl di nota normalità.
Reagenti necessari
:HCl N/10 f.c. noto Metilarancio
Modo di operare:
DUREZZA TEMPORANEA (valida in assenza di carbonati alcalini)
Si prelevano 250 ml esatti di acqua in esame, si portano in un beaker, si aggiungono 3 gocce di metilarancio e si titola con HCl N/10 f.c. noto fino a viraggio rosa.
P. eq. CaCO3 = (PM CaCO3 : 2) = 50,045
N.B.: I g di CaCO3 (anche il Mg è valutato come Ca), rapportati a 100 litri, rappresentano i gradi di durezza francesi. Oppure:
P. eq. CaO = (PM CaO : 2) = 28,04
N.B.: I g di CaO (anche il Mg è valutato come Ca), rapportati a 100 litri, rappresentano i gradi di durezza tedeschi.
La durezza temporanea e totale di un'acqua, viene però più vantaggiosamente determinata col metodo complessometrico (vedi pag. 53).
DETERMINAZIONE DELL'ALDEIDE FORMICA
Reagenti necessari:
NaOH N/10 f.c. noto H2O2 a 10 - 12 vol
HCl N/10 c. noto Fenolftaleina
Modo di operare:
Grammi 2 (oppure ml 2) di formaldeide commerciale in soluzione, vengono portati in matraccio tarato al volume di 250 ml con acqua distillata. Si prelevano 25 ml di tale soluzione, si portano in un beaker e vi si aggiungono 50 ml esatti di NaOH N/10 f.c. noto, e 4-5 ml di acqua ossigenata H2O2 a 10-12 vol. Si scalda debolmente per 20' e si agita per scacciare l'ossigeno in eccesso, quindi si diluisce e si titola con HCl N/10 f.c. noto fino a scoloramento della fenolftaleina.
(50 ml di NaOH N/10 x f.c.) – (ml usati di HCl N/10 x f.c.) = ml corrispondenti alla formaldeide.
1.000 : 30,026/10 = ml risultati : X
X . 10 = g di formaldeide presenti in 2 g (o 2 cc) di campione
X . 10 – 50 = % in peso o in volume a seconda che si sono prelevati 2 g o 2 ml
L'aldeide formica commerciale è generalmente al 30-40% (formalina); l'acqua ossigenata H2O2 la ossida, in soluzione basica, ad acido formico:
H-CHO + H2O2 + NaOH þ H-COONa + 2 H2O
Il formiato di sodio che si forma in seguito alla titolazione, ha un pH circa 8 (l'acido formico è più forte dell'acetico) e i suoi alcalini non arrossano la fenolftaleina; così, si può titolare l'Na0H in eccesso, unica responsabile dell'arrossamento della fenolftaleina.
Questa determinazione, che riguarda i sali solubili di mercurio-ico, si basa sulla precipitazione del mercurio come ossido con idrato sodico o potassico in eccesso noto e sulla titolazione dell'eccesso di alcali con acido cloridrico titolato.
La precipitazione del mercurio come ossido è accompagnata da un trattamento con acqua ossigenata allo scopo di trasformare l'ossido in mercurio metallico che non ha alcuna azione sull'acido cloridrico usato nella titolazione. Le reazioni sono:
HgCl2 + 2 NaOH þ HgO + 2 NaCl + H2O
HgO + H2O2 þ Hg + H2O + O2
Reagenti necessari:
Acqua ossigenata 3% (10 vol) neutra NaOH N/10 HCl N/10
Fenolftaleina
Modo di operare:
Un sale solubile di Hg-ico in quantità di circa g 0,3 -di Hg viene sciolto in 100-200 ml di acqua distillata neutralizzando, se occorre la soluzione risultante alla fenolftaleina; si aggiungono 5 ml di acqua ossigenata a 10 vol (a sua volta neutralizzata, se occorre, alla fenolftaleina) e quindi a caldo lentamente ed agitando 50 ml di NaOH N/10 f.c. noto; si tiene il tutto sotto ebollizione per 15' ed il mercurio si deposita completamente allo stato di metallo grigio. Si aggiungono quindi 50 ml di HCl N/10 f.c. noto. Si fa bollire ancora 5' e si titola con NaOH N/10 f.c. noto alla fenolftaleina. Indicando con x i ml usati in quest'ultima titolazione avremo:
(50 + x) . f.c. NaOH - 50 . f.c. HCl = ml corrispondenti al Hg
p. eq. Hg/2 = 100,3 p. eq. HgCl2 = HgCl2 /2 = 135,76
GENERALITA' - PREP. SOLUZ. NITRATO D'ARGENTO N/10 E DET. DEL FATT. CORR. (SOST. BASE: CLORURO DI SODIO) - PREP. SOLUZ. SOLFOCIANURO AMMONICO N/10 E DET. FATT. CORR. - DET. Cl- MOHR (Br- e I-) - DET. 'CI VOLHARD (Br-, I-, CN-, CNS-) - DET. CN-.
L'argentometria è un metodo di analisi volumetrica per precipitazione e si basa sull'uso di soluzioni di AgNO3, a contenuto noto, approfittando del fatto che molti anioni sono ben precipitabili come sali d'argento.
A pag. 2 già si è trattato in generale sulla teoria della precipitazione frazionata dei sali d'argento, teoria che sarà applicata ai vari casi qui di seguito descritti.
PREPARAZIONE DELLA SOLUZIONE DI AgNO3 N/10 E DETERMINAZIONE DEL SUO FATTORE CORRETTIVO (sostanza base cloruro di sodio)
Il peso equivalente del AgNO3 = PM AgNO3 = 169,888. Una sua soluzione N/10 dovrà perciò contenere g 16,9888 di AgNO3 puro per litro.
Modo di operare
:Pesare perciò alla bilancia analitica g 17 di AgNO3 (leggero eccesso per eventuali impurità), portarli in un matraccio tarato da 1000 ml e portarli a volume con acqua distillata dopo aver ottenuto la completa soluzione del sale.
La soluzione di AgNO3, così preparata, dovrà essere controllata mediante una prova in bianco che viene eseguita su una quantità esattamente pesata di NaCl purissimo e secco. Si determina cioè il suo fattore correttivo nel seguente modo.
Modo di operare:
Si pesa esattamente una quantità di NaCl purissimo prossima a g 0,15 in un vetrino e si porta in un beaker sciogliendola in 50-100ml di acqua distillata; si aggiunge un ml di cromato di K2CrO4 in soluzione all'8% che funge da indicatore.
Si titola con la soluzione di AgNO3, N/10 in esame calata da una buretta fino a viraggio leggermente rosso persistente per 5’.
I ml usati in tale determinazione, vengono denominati ml pratici.
Si esegue ora, il calcolo teorico sulla quantità esattamente pesata Q di NaCl purissimo:
1.000 : 5,8454 = X :Q (5,8454 = p. eq. NaCl N/10) X = ml teorici
ml teorici / ml pratici = fattore correttivo
La soluzione di AgNO3, preparata, avrà quindi la seguente denominazione:
AgNO3 N/10 f.c. . .....
Ogni qualvolta si userà questa soluzione, bisognerà moltiplicarne i ml usati per tale fattore.
Il meccanismo della titolazione e del viraggio, consistono nella precipitazione frazionata fra il AgNO3, introdotto ed il NaCl e K2CrO4 presenti nel beaker infatti l'AgNO3 reagisce con tutto l'NaCl:
AgNO3 + NaCl þ NaNO3 + AgCl prec. bianco
L'ulteriore goccia di AgNO3, reagisce con l'indicatore K2CrO4:
2 AgNO3+ K2CrO4 þ 2 KNO3 + Ag2CrO4 prec. rosso
Il cromato d'argento Ag2CrO4 rosso, si forma soltanto dopo che tutto il Cl- è stato precipitato come AgCl; infatti quando l'AgCl (P = [Ag+] [Cl-] = 10-10) è stechiometricamente precipitato e presente come corpo di fondo (punto di equivalenza), la conc. Ag+ sovrastante sarà,
[Ag+] = ![]() =
10-5 cioè sufficiente per iniziare la formazione del Ag2CrO4
=
10-5 cioè sufficiente per iniziare la formazione del Ag2CrO4
(P = [Ag+]2 [CrO4--]
= 10-12) [Ag+] =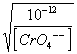 = 10-5
= 10-5
(ammettendo indicativamente che [CrO4--] = 10-2).
Si tratta dunque di una precipitazione frazionata nella quale precipita per primo il AgCl e poi il Ag2CrO4 con l'ulteriore goccia.
PREPARAZIONE DELLA SOLUZIONE DI NH4CNS N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO
Il peso equivalente del solfocianato ammonico NH4CNS è 76,118 e perciò, una soluzione N/10 dovrà contenere g 7,6118 nel litro.
Il NH4CNS è una sostanza piuttosto igroscopica: sarà bene perciò, in una preparazione approssimata, pesarne una quantità superiore.
Modo di operare
:Pesare in un vetrino circa g 8 di NH4CNS e portarli in un matraccio tarato da 1000 ml.
Portare a volume con acqua distillata.
La soluzione di NH4CNS così preparata, viene controllata sulla soluzione di AgNO3 N/10 f.c. noto nel seguente modo:
Modo di
operare:In un beaker, si pongono 20 ml esattamente misurati con una pipetta di AgNO3 /10 f.c. noto, si diluiscono a circa 80 -100 ml con acqua distillata e si aggiunge un ml di allume ferrico-ammonico
(NH4)2 SO4 . Fe2 (SO4)3 .24 H2O in soluzione acquosa satura acidificata con un ml di HNO3 conc. per 100 ml di soluzione.
Si titola tale soluzione col NH4CNS calato da una buretta graduata, fino ad ottenere una lieve colorazione rossa persistente per 2 minuti.
I ml usati in tale operazione vengono denominati ml pratici.
I 20 ml di AgNO3 moltiplicati per il suo f.c. noto, rappresentano i ml teorici.
ml teorici / ml pratici= fattore correttivo
La soluzione di NH4CNS preparata, avrà perciò la seguente denominazione:
NH4CNS N/ 10 f.c. . .....
Ogni qualvolta si userà tale soluzione, bisognerà moltiplicarne i ml usati per tale f.c.
Il meccanismo di questa titolazione e del viraggio, si basa sulla reazione fondamentale fra AgNO3, e NH4CNS:
NH4CNS + AgNO3 þ NH4NO3 + AgCNS prec. bianco
Esauriti tutti gli ioni Ag+, l’ulteriore goccia di NH4CNS reagisce coll'indicatore allume ferrico secondo la reazione:
Fe+++ + CNS- þ Fe(CNS)++ coloraz. rosso sangue
La colorazione rossa indica la fine della precedente reazione.
Lo ione colorato Fe(CNS) ++ si formerà assolutamente dopo la precipitazione totale del AgCNS; infatti quest'ultimo ha un Pr. di sol. [Ag+] . [CNS-] = 10-12 e quando sarà presente come corpo di fondo stechiometrico al punto di equivalenza della titolazione, la conc. CNS- sarà [CNS-] = V 10-12 = 10-6 che è proprio la concentrazione sufficiente ad iniziare la formazione dello ione colorato Fe(CNS)++ che quindi si formerà, con l'ulteriore goccia, in quantità sufficiente da rendere visibile la colorazione.
DETERMINAZIONE DELLO IONE Cl- (acido cloridrico e cloruri) SECONDO MOHR
(idem per Br- e I-)
Viene eseguito mediante titolazione diretta con AgNO3, di normalità nota su una soluzione neutra
contenente i Cl- e K2CrO4 quale indicatore.
La neutralità della soluzione, ha fondamentale importanza poichè, in soluzione basica si avrebbe
precipitazione di ossido di argento Ag2O ed in soluzione acida non si formerebbe il Ag2CrO4 rosso (che è solubile negli acidi) che indica la fine della titolazione.
Il pH è compreso fra 5 e 10; se la soluzione è acida, viene neutralizzata con bicarbonato sodico;
invece se la soluzione è basica viene neutralizzata con acido acetico diluito.
Si opera come segue:
Reagenti necessari:
Ag NO3 N/10 f.c. noto K2CrO4 al 8% cartine da pH 5-10
Modo di operare:
La soluzione in esame (contenente Cl- in quantità prossima all'equivalente di 30-40 ml di
soluzione N/10) va controllata alla tocca su cartine da pH e deve risultare fra 5 e 10; diversamente si corregge con NaHCO3, oppure con gocce di CH3COOH dil. controllando ancora il pH.
Si aggiunge un ml di K2CrO4, in soluzione aq. 8% e si titola con AgNO3 f.c. noto calato da una
buretta, fino a viraggio lievemente rosso.
(Per valutare esattamente il punto di viraggio, si prepara un volume d'acqua pari al titolato e vi si
aggiunge un ml di indicatore K2CrO4; si calano su tale soluzione le gocce di AgNO3 N/10 f.c. noto necessarie per ottenere una colorazione pari a quella della soluzione del Cl- esaminata e, queste gocce, vengono dedotte dalla lettura precedente).
I ml usati e corretti di AgNO3 N/10 f.c. noto corrispondono ai Cl- presenti.
1.000 : p. eq. Cl- (Br-, I-) / 10 = ml usati e corretti : X
P.eq. Cl- = (PA Cl) = 35,457
P.eq. Br- = (PA Br) = 79,916
DETERMINAZIONE DEL Cl-, Br-, I-,CN-,CNS- SECONDO VOLHARD
Questa determinazione, si basa sull'aggiunta alla soluzione contenente gli ioni Cl-, Br-, I-,CN-,CNS- di una quantità esatta ed un eccesso di AgNO3 di normalità nota ottenendo la precipitazione totale dello ione Cl- (id.); l'eccesso di AgNO3, viene a sua volta titolato con un soluzione di NH4CNS di nota normalità usando allume ferrico quale indicatore.
Avremo cioè:
Aggiunta di AgNO3 misurato in eccesso:
1) XCl + AgNO3 þ XNO3 + AgCl
Titolazione dell'eccesso di AgNO3 con NH4CNS:
2) AgNO3 + NH4CNS þ NH4N03 + AgCNS
Azione del NH4CNS sull'indicatore ferrico:
3) Fe+++ + CNS- þ Fe(CNS)++ colorazione rosso sangue
La colorazione retrocede lentamente per azione dei pur pochi ioni Ag+ che tendono ad assorbire ioni CNS- del Fe(CNS)++ per formare AgCNS, che fra i precipitati è il meno solubile e perciò si forma anche a spese degli altri.
4) AgCl + Fe(CNS)++ þ AgCNS + Fe+++ Cl-
P = 10-10 rosso P = 10-12 incolore
Tutta l'operazione viene condotta in soluzione acida per HNO3 esente da HNO2; infatti il HNO2 produce, sull'allume ferrico, una colorazione rossa che interferisce sul viraggio. Per eliminare l'eventuale HNO2 presente nell'HNO3 basta bollire quest'ultimo sotto cappa per pochi minuti.
Reagenti necessari:
AgNO3 N/10 f.c. noto NH4CNS f.c. noto
Allume ferrico soluz. satura HNO3 conc.
Modo di operare:
La soluzione in esame (contenente Cl- in quantità prossima all'equivalente di 30-40 ml di soluzione N/10), viene portata a 100 ml circa con acqua distillata e acidificata con 1 ml di HNO3 conc. (esente da HNO2).
A tale soluzione, si aggiungono 50 ml esatti, di AgNO3, f.c. noto ottenendo un precipitato bianco di AgCl.
Dopo raffreddamento si aggiunge un ml di allume ferrico (NH4)2SO4 - Fe2(SO4)3 - 24H2O in soluzione aq. satura e si titola con NH4CNS f.c. noto calato da una buretta graduata, fino a colorazione lievemente rossa ancorchè poco persistente.
Il calcolo si esegue nel seguente modo:
50 ml di AgNO3 N/10 opportunamente corretti meno i ml di NH4CNS N/ 10 opportunamente corretti usati nella titolazione, danno i ml corrispondenti al Cl- da determinare:
P. eq. Cl = (PA Cl) = 35,457
P. eq. Br = (PA Br) = 79,916
P. eq. I = (PA I) = 126,92
P. eq. CN = (PA CN) = 26,018
P. eq. CNS = (PA CNS) = 58,078
DETERMINAZIONE DELLO IONE CN- (acido cianidrico e cianuri)
Questa determinazione, si basa sulla formazione dello ione complesso Ag(CN)2- e successiva decomposizione.
Infatti, il AgNO3, calato in una soluzione alcalina contenente ioni CN- forma lo ione complesso argento cianuro Ag(CN)2- solubile (il temporaneo precipitato di Ag2(CN)2 nel punto dove cade la goccia, si dissolve immediatamente nell'eccesso dei CN-). Esauriti tutti i CN-, l'ulteriore goccia di AgNO3, introdotta, produce la precipitazione dell'argento cianuro d'argento Ag[Ag(CN)2] ovvero Ag2(CN)2 insolubile, che forma perciò un intorbidamento bianco stabile che segna la fine della reazione.
2AgNO3 + 2NaCN þ 2NaNO3 + Ag2(CN)2 prec. bianco temporaneo
Ag2(CN)2 + 2NaCN þ 2NaAg(CN)2 argento cianuro di sodio solubile
2NaAg(CN)2 + AgNO3 þ NaNO3 + Ag2(CN)2 prec. bianco definitivo
Naturalmente, in questo caso, il peso equivalente del CN- sarà il doppio del peso CN-, in quanto ad un atomo di Ag corrispondono due radicali CN nello ione complesso che si forma Ag(CN)2-.
In presenza di sali ammonici, e cioè di ammoniaca data l'alcalinità della soluzione, non si può cogliere il viraggio perchè il precipitato definitivo di Ag2(CN)2 si scioglie dando il complesso ammoniacale
Ag2(CN)2 + 4 NH3 þ 2 Ag(NH3)2CN solubile.
In tal caso, si aggiunge una piccola quantità di KI, il quale funziona da indicatore per intorbidamento perché il AgI che si forma alla fine non è solubile in NH3.
Reagenti necessari:
AgNO3 N/10 f.c. noto cartine da pH 7-9
Modo di operare:
La soluzione contenente un esatto peso di campione calcolato preventivamente in forma approssimata
intorno all'equivalente in CN (2 x 26) di 30-40 ml di soluzione N/10, deve essere alcalina sul pH 7-9 (controllare con le cartine da pH) ed esente da sali ammonici. Si titola con AgNO3 N/10 f.c. noto calato da una buretta, agitando continuamente la soluzione, fino a persistente intorbidamento bianco.
I ml usati e corretti, corrispondono al CN- il cui peso eq. è in questo caso doppio.
P. eq. CN = (2 x PA CN) = 52,036
Sotto il nome generico di ossidimetria viene indicato un metodo di analisi volumetrica basata su reazioni di ossidoriduzione; la permanganatometria, iodometria, bromatometria, ne sono casi particolari.
Le reazioni di ossidoriduzione
sono reazioni che prevedono una variazione di valenza nelle sostanze partecipanti ossia un trasferimento di elettroni da un elemento ad un altro. Il trasferimento elettronico segue la via di una caduta di potenziale e precisamente si verifica dall'elemento più alto al più basso nella serie elettrochimica.
Serie elettrochimica per la parte che interessa l'ossidimetria:
elettroni trasferiti
Sn++ / Sn++++ E° = + 0, 15 Volt 2
Cu+ / Cu++ + 0,16 1
4 6
SO3-- / SO4-- + 0,17 2
OH- / ½ H2O . ¼ O2 + 0,40 1
(COO-)2 / 2 CO2 + 0,49 2
(S2O3--) / ½ (S4O6--) + 0,50 1
0 1
½ Cl2 / (ClO-) + 0,12 1
I- / ½ I2 + 0,53 1
3 5
(AsO3 - - - ) / (AsO4---) + 0,56 2
Fe++ / Fe+++ + 0,77 1
Hg+/Hg++ + 0,91 1
2 5
(NO) / (NO3 -) + 0,95 3
Br- / ½Br2 + 1,09 1
I- / (IO3-) + 1,2 6
Cr+++ / (CrO4--) + 1,30 3
Cl- / ½Cl2 + 1,35 1
0 5
½ Br2 / (BrO3-) + 1,48 5
2 7
Mn++ / (MnO4-) + 1,52 5
3 5
Bi+++ / (BiO3-) + 1,7 2
(SO4-- / (½ S2O8) + 1,90 1
Il potenziale normale E°, secondo il quale la serie elettrochimica è redatta, rappresenta il potenziale minimo al quale gli elettroni possono essere acquistati o ceduti da un elemento ed indica l'affinità elettronica; quindi in coda alla serie si ritrovano gli ossidanti (accettori di elettroni) ed in testa i riducenti (datori di elettroni); ad ogni modo, ogni elemento è ossidante rispetto a quelli che gli stanno sopra e riducente rispetto a quelli che gli stanno sotto; per es.: il cromato di potassio K2CrO4 è ossidante rispetto al sali ferrosi ma è meno ossidante del permanganato che anzi è capace di ossidare i sali di Cr+++ a cromati CrO4--.
Esaminiamo ora, come esempio, la reazione di ossidoriduzione fra cloruro stannoso e cloruro mercurico:
SnC12 + 2HgCl2 þ SnCl4 + Hg2Cl2
in forma ionica Sn++ + 2Hg++ þ Sn++++ + 2Hg+
in forma elettronica 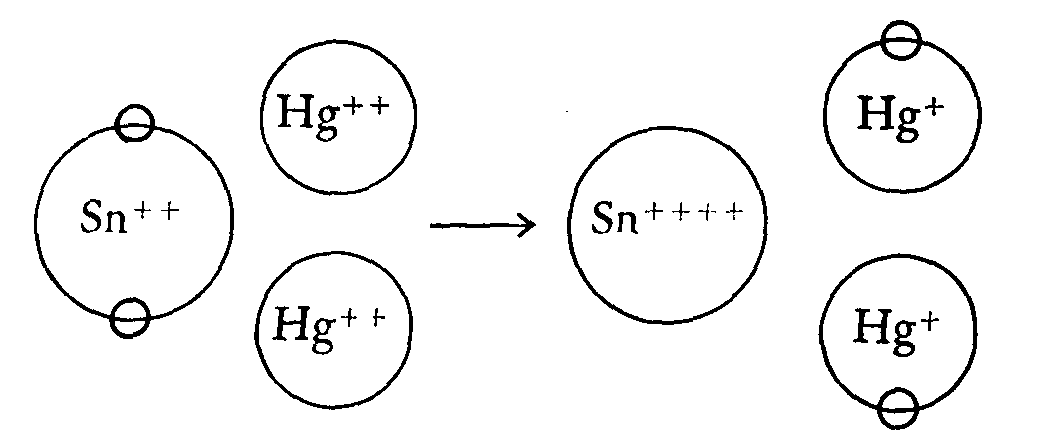
Si vede che gli elettroni si sono trasferiti dallo Sn++ che diventerà Sn++++ al 2Hg++, che diventeranno 2Hg+; le reazioni, di fatto, sono due concatenate:
- 2e +2e
Sn++ þ Sn++++ 2 Hg++ þ 2 Hg+
Nella prima, lo Sn++ riducente, si ossida; nella seconda, contemporaneamente, il Hg++ ossidante, si riduce.
In definitiva sono riducenti tutti quegli elementi che in una reazione perdono elettroni (ed acquistano di valenza) mentre sono ossidanti tutti quegli elementi che acquistano elettroni (e perdono di valenza); il trasferimento elettronico avviene sempre dall'alto al basso della serie elettrochimica verso i potenziati più positivamente elevati che quindi indicano l'affinità elettronica.
Si definisce potenziale di ossidoriduzione di una reazione, la ddp esistente fra i due elementi che reagiscono e, più elevata è tale ddp e più spinta e quantitativa sarà la reazione.
E = E° Hg+/Hg++ - E°Sn++/Sn++++ = 0,91 - 0,15 = 0,76 V
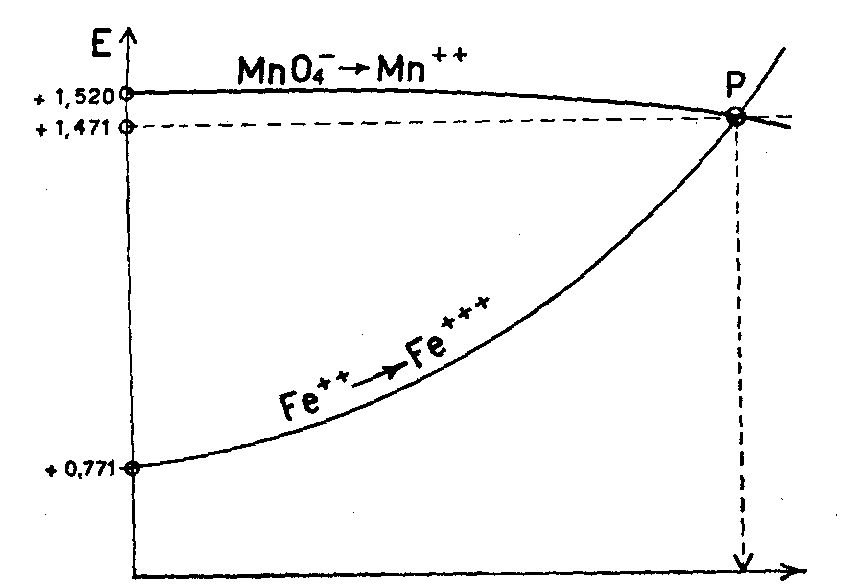
2. Fra permanganato e sali ferrosi, il potenziale di ossidoriduzione sarà:
E = E° Mn2+(MnO4-)7+ - E° Fe++/Fe+++ = 1,52 - 0,77 = 0,75 V
Nel caso che in soluzione siano possibili diverse reazioni di ossidoriduzione, si verifica sempre per prima quella che presenta il potenziale di ossidoriduzione maggiore, cioè reagiscono per primi i due elementi più lontani nella serie. Così, se per esempio, caliamo da una buretta del KMnO4 su una soluzione contenente Fe++ acida per HCl, dapprima il KMnO4 (E° = + 1,52) ossida il Fe++ a Fe+++ (E° = + 0,77) e poi ossida lo ione Cl- a cloro (E° = + 1,35).
Naturalmente la ddp esistente all'inizio di una reazione di ossidoriduzione va gradatamente estinguendosi perchè la reazione stessa produce una variazione nella concentrazione dei vari ioni (nell'esempio del SnCl2 + 2HgCl2 diminuiscono le conc. Sn++ e Hg++ ed aumentano le conc. Sn++++ e Hg+) che hanno l'effetto di aumentare il potenziale più basso e diminuire il potenziale più alto fino a renderli eguali; in tal punto, la ddp sarà nulla e la reazione è definitivamente spenta.
Il diagramma qui a fianco riportato indica l’andamento, del potenziale del KMnO4 e dello ione Fe++ durante una titolazione e si vede come al punto P la reazione sia finita. P è il punto di equivalenza della reazione.
GENERALITA' - PREP. SOLUZ. PERMANGANATO DI POTASSIO N/10 E DET. FATT. CORR. (SOST. BASE: OSSALATO SODICO) - DET. Fe ZIMMERMANN - DET. Ca (E OSSALATI) - DET. ACQUA OSSIGENATA (PERBORATI E PERCARBONATI) DET. DEL Cr (AL PERSOLFATO) - DET. DELLO Sn.
Il Permanganato di potassio KMnO4, e una sostanza cristallina violetta che agisce da ossidante sia in ambiente basico che neutro che acido;
2 KOH 7 + 6+
2 KMnO4 þ 2 K2MnO4 + H2O + 1/2 O2 (MnO4-/MnO4--)
2. in ambiente neutro o debolmente basico è un ossidante trivalente e si riduce a biossido di manganese:
H2O 7+ 4+
2 KMnO4 þ 2 MnO2 + 2 KOH + 3/2 O2 (MnO4-/MnO2)
3. in ambiente acido è un ossidante pentavalente e si riduce a sale manganoso:
3 H2SO4 7+ 2+
2 KMnO4 þ 2 MnSO4 + K2SO4 + 3 H2O + 5/2 O2 (MnO4-/Mn++)
Le tre equazioni chimiche scritte qui sopra servono soltanto ad indicare il potere ossidante del KMnO4 nei vari ambienti, ma non sviluppano ossigeno di per se; l'ossigeno indicato rappresenta quantitativamente la capacita ossidante rispetto a sostanze riducenti la cui presenza, è assolutamente necessaria al verificarsi delle reazioni.
Il KMn04 è usato in soluzione acida e presenta il particolare vantaggio di decolorarsi (MnO4- = viola; Mn++ = rosa chiaro) man mano che reagisce e di non aver quindi bisogno di indicatori.
Il suo Peso equivalente è (PM:5 = 158,03:5 = 31,605), e per preparare una soluzione N/10 sono necessari g 3,1605 per litro.
PREPARAZIONE DELLA SOLUZIONE DI KMnO4 N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO (sostanza base ossalato sodico)
Modo di operare:
Su un vetrino precedentemente tarato, si pesano g 3,17 (leggero eccesso per via delle impurità) di KMnO4 cristallino, si trasportano in un matraccio tarato da 1000 ml e si porta a volume con acqua distillata.
Si lascia a se per qualche giorno affinché il KMnO4 si stabilizzi di fronte alle impurità riducenti dell'acqua.
La soluzione così preparata, deve essere controllata mediante una prova in bianco su una quantità esattamente nota di ossalato sodico (COONa)2 puro e seccato in stufa a 100° C per due ore.
L'azione del KMnO4 sull'ossalato sodico è la seguente:
2 KMnO4 + 5 (COONa)2+ 8 H2SO4 þ K2SO4 + 2 MnSO4 + 5 Na2SO4 + 8 H2O + 10 CO2
L'ossalato sodico, ha numero dì riduzione due come risulta dal suo rapporto molecolare col KMnO4.
Il potenziale di ossidoriduzione normale di questa reazione è E° KMnO4 - E° ossalato = 1,52 - 0,49 = 1,03 volt cioè la reazione è notevolmente spinta.
Allo scopo di determinare il f.c. della soluzione di KMnO4 N/10 preparato, si procede nel seguente modo:
Modo di operare:
Si pesa esattamente una certa quantità di ossalato sodico puro e secco, prossima all'equivalente di 30-40 ml di soluzione N/10 e cioè g 0,2-0,25 (P. eq. Ossalato sodico = PM : 2 = 67,007).
Si porta tale quantità in un beaker e vi si aggiungono 100 ml di acqua distillata e 20 ml di H2SO4 díl. (10% circa); si scalda sui 70° C e si titola col KMnO4 N/10 in esame calato da una buretta, fino a che la colorazione rosa del KMnO4 persiste stabilmente.
I ml usati in tale titolazione, vengono denominati ml pratici.
Invece dall'ossalato sodico pesato Q, si deducono i ml teorici:
1.000 : 6,7007 = X : Q
ml teorici / ml pratici = fattore correttivo
La soluzione preparata, avrà perciò la denominazione:
KMnO4 N/10 f.c. .....
Ogni qualvolta sarà usata tale soluzione, bisognerà moltiplicare i ml usati per il f.c.
DETERMINAZIONE DEL Fe++ E DEL Fe+++ SECONDO ZIMMERMANN
Questa determinazione si basa sull'azione ossidativa del KMnO4, sui sali ferrosi Fe++. E' pertanto assolutamente necessario che la soluzione esaminata contenga il ferro sotto forma di Fe++ . Se invece il ferro fosse presente come ferrico Fe+++, esso deve essere preventivamente ridotto a ferroso Fe++ operando nel modo che verrà indicato più avanti.
Il permanganato di potassio KMnO4, agisce sui sali ferrosi in soluzione secondo la seguente reazione:
2 KMnO4 + 10 FeSO4 + 8 H2SO4 þ K2SO4 + 2 MnSO4 + 5 Fe2(SO4)3 + 8 H20
Dal rapporto molecolare oppure dal salto del numero di ossidazione Fe+ + þ Fe +++ si deduce che il Fe vale 1, ed i1 suo peso atomico coincide con l'equivalente.
Il potenziale di ossidoriduzione normale di questa reazione è E°KMnO4 - E°Fe = 1,52 - 0,77 = 0,75 volt cioè la reazione è abbastanza spinta.
Per favorire tale reazione e per renderne più evidente il viraggio (persistenza della colorazione rosa del KMnO4), si opera in presenza della soluzione di Zimmermann che contiene solfato manganoso MnSO4, acido fosforico H3PO4 e acido solforico H2SO4:
Si opera come segue:
Reagenti necessari:
MnSO4 . 4 H2O H3PO4 d = 1,7 H2SO4 d = 1,8Modo di operare:
Preparazione della soluzione Zimmermann:
Si pesano g 67 di MnSO4 . 4H2O, si portano in un beaker da 800-1000 ml e si sciolgono in 500 ml di acqua distillata; si aggiungono 100 ml di acido fosforico d = 1,7 e quindi 100 ml di acido solforico d = 1,8. La soluzione, agitata e stabilizzata, viene portata, in un recipiente da un litro, a volume con acqua distillata.
Determinazione del ferro ferroso Fe++:
Un campione di sale ferroso, (contenente Fe++ in misura approssimativamente equivalente a 30-40 ml di soluzione N/10) (p. eq. Fe = 55,84), viene portato in 100 ml di soluzione acida per HCl o H2SO4. Vi si aggiungono circa 10 ml di soluzione Zimmermann e si titola a freddo con KMnO4 N/10 f.c. noto calato da una buretta fino a colorazione rosa persistente.
I ml usati e corretti, rappresentano il Fe.
P. eq. Fe = (PA Fe) = 55,84
Se invece il ferro è presente come ione ferrico Fe+++, si rende necessaria la sua riduzione che può essere eseguita con zinco in soluzione acida (sviluppo di idrogeno riducente H2), con anidride solforosa SO2, oppure con cloruro stannoso SnCl2.
In quest'ultimo caso, il SnCl2 agisce nel seguente modo:
2 FeCl3 + SnCl2 þ 2 FeCl2 + SnCl4 (cloruro stannico)
Il SnCl2 aggiunto in leggero eccesso, deve essere distrutto; diversamente ridurrebbe il KMnO4 alterando i risultati dell'analisi. Per distruggerlo, si aggiunge cloruro mercurico HgCl2:
SnCl2 + 2 HgCl2 þ SnCl4 + Hg2Cl2 (cloruro mercuroso insolubile ed inattivo di fronte al KMnO4)
L'eccesso di SnCl2 dove essere il minimo possibile per evitare che il cloruro mercurico venga ridotto, non soltanto a mercuroso, ma addirittura a Hg nero (Hg2Cl2 + SnCl2 þ SnCl4 + 2 Hg nero), nel qual caso bisogna ripetere l'analisi usando minor quantità di SnCl2.
Eseguita la riduzione, si titola il Fe++.
Si opera nel seguente modo:
Reagenti necessari
:SnCl2. 2 H2O HCl conc. HgCl2 soluz. Zimmermann KMnO4 N/10 f.c. noto.
Modo di operare
:Riduzione del ferro ferrico Fe+++ a ferroso Fe++ e determinazione.
Anzitutto, si preparano le soluzioni di SnCl2 e HgCl2 rispettando le seguenti indicazioni:
soluzione di cloruro stannoso: 250 g di SnCl2 . 2H2O si introducono un po' alla volta in un bottiglione da 2 litri contenente una soluzione acida preparata con 1000 ml di H2O dist. e 300 ml di HCl conc.; ottenuta la soluzione si porta al volume di 2 litri con acqua distillata. Introdurre qualche pezzo di stagno metallico per la conservazione.
soluzione di cloruro mercurico: soluzione satura di HgCl2 (5-6%).
Procedimento: il sale ferrico, in una piccola beuta in soluzione il più possibile concentrata e fortemente cloridrica viene portato all'ebollizione. Si aggiunge goccia a goccia e lentamente la soluzione di cloruro stannoso a decolorazione e poi ancora qualche goccia. Tale soluzione decolorata, diluita al triplo con acqua dist., viene lentamente versata su un'altra soluzione del volume di 200 ml contenenti 10 ml di soluzione HgCl2 lavando bene la beuta contenente la prima soluzione e versando, nella seconda soluzione le acque di lavaggio. Si lascia a se per qualche minuto che si formi il precipitato bianco di Hg2Cl2 (Se si forma precipitato nero, bisogna riprendere tutta la riduzione usando meno cloruro stannoso(COONa)2.
Si aggiungono 10 ml. di soluzione Zimmermann e si titola con KMnO4 N/10 f.c. noto come detto per la determinazione dei sali ferrosi.
DETERMINAZIONE DEL CALCIO (E DEGLI OSSALATI)
In questa determinazione, il Ca viene precipitato con ossalato ammonico (COONH4)2
come ossalato di calcio bianco cristallino:
CaCl2 + (COONH4)2 þ 2 NH4Cl + (COO)2Ca precipitato bianco di ossalato di calcio
L'ossalato di calcio, filtrato e lavato, viene trattato con acido solforico, mettendo in libertà l'acido ossalico:
(COO)2 Ca + H2SO4 þ CaSO4 + (COOH)2 acido ossalico
L’acido ossalico, che è evidentemente equivalente al calcio da determinare (1 mol. di acido ossalico per ogni atomo di Ca), viene titolato con KMnO4 di normalità nota secondo la seguente equazione:
2 KMnO4 + 5 (COOH)2 + 3 H2SO4 þ K2SO4 + 2 MnSO4 + 10 CO2 + 8 H2O
L'equivalente del Ca sarà pari a quello dell'acido ossalico, e cioè (PA Ca : 2) = 20,04.
Si opera come segue:
Reagenti necessari:
KMnO4 N/10 f.c. noto NH4Cl Fenolftaleina NH3 al 10%
(COONH4)2 al 5 % AgNO3 5% H2SO4 al 10%
Modo di operare:
Il campione contenente Ca (in quantità approssimativamente equivalente a 30-40 ml di soluzione N/10), viene portato in soluzione leggermente cloridrica 50-100 ml totali).
Si aggiunge 1-2 g di cloruro ammonico NH4Cl solido, 3 gocce di fenolftaleina ed NH3 diluito fino a colore rosa. Si porta tutto all'ebollizione e si aggiunge goccia a goccia e continuamente agitando, una soluzione bollente di ossalato ammonico (soluzione al 5% circa) fino a completa precipitazione; si aggiungono ancora 10 ml di ossalato ammonico, si copre il beaker con un vetro da orologio e si lascia a se per 12 ore affinché i piccoli cristalli di ossalato di calcio ingrossino.
Si decanta il liquido sovrastante attraverso un filtro a fascia bianca e si lava il precipitato nel beaker tre volte per decantazione con soluzione 0,1 % di ossalato ammonico.
Si porta il precipitato sul filtro dove viene lavato in tre volte con acqua distillata.
Il filtrato non deve dare la reazione del Cl- con AgNO3 in soluzione nitrica nè, soprattutto, la reazione dello ione ossalico con KMnO4 diluitissimo in soluzione solforica (decolorazione); se il filtrato desse una delle reazioni predette, necessita un ulteriore lavaggio perchè sia lo ione Cl- che l’ossalico ridurrebbero il KMnO4 di titolazione, portando un errore in più).
Si pone sotto l'imbuto un beaker pulito e, forando il filtro con una bacchetta, si trasporta il precipitato nel sottostante beaker trattandolo con acqua calda; si lava molto bene il filtro con H2SO4 al 10% caldo fino ad usarne circa 100 ml ed ottenendo nel beaker una soluzione limpida.
Allora si porta il beaker sotto la buretta del KMnO4 N/10 f.c. noto e si titola fino a colore rosa persistente.
P. eq. Ca = (PA Ca : 2) = 20,04
DETERMINAZIONE DELL'ACQUA OSSIGENATA (E DEI PERCARBONATI E PERBORATI)
L’acqua ossigenata H2O2, reagisce col KMnO4 in soluzione solforica secondo la seguente equazione:
2 KMnO4 + 5 H2O2 + 3 H2SO4 þ K2SO4 + 2 MnSO4 + 5 O2 + 8 H2O
Esaurita l'H2O2, il KMnO4 rimane indecomposto e colora in rosa la soluzione.
Il p.eq. del l'H2O2 (H2O2 þ H2O + O) e (PM H2O2 : 2) = 17,008 che, fra l'altro corrisponde a 1/2 atomo di ossigeno libero, cioè ad 1/4 di molecola (O2) e cioè come gas, a ml (22,414 : 4) = ml 5603 c.n.
Infatti, il titolo dell'acqua ossigenata, può essere dato in % (g di H2O2 per 100 ml) oppure in volumi e cioè i ml di ossigeno gas sviluppati da un ml di soluzione di H2O2.
Si opera nel seguente modo:
Reagenti necessari:
KMnO4 N/10 f.c. noto H2SO4 al 20%
Modo di operare:
Il campione, deve equivalere a circa 30-40 ml di soluzione N/10 che corrispondono appunto a circa g 0,05 di H2O2 (1.000: 1,7008 = 30 : X) ovvero a circa 17 ml di ossigeno libero (1.000 : 5603 = 30 : X). Così, opinando che il titolo dell'acqua ossigenata in esame sia p% oppure v volumi, i ml da prelevare ed esaminare saranno prossimi ai valori:
0,05 . 100 / P ovvero 17 / v
Il prelievo potrà essere anche 10 o 20 volte superiore a quello preventivato, quindi diluito ad opportuno ed esatto volume con acqua distillata, operando infine sull'opportuna aliquota.
La soluzione acquosa contenente l'acqua ossigenata in un volume di 70-100 ml, viene aggiunta di 20 ml di H2SO4 al 20% e titolata con KMnO4 N/10 f.c. noto fino a persistente colore rosa.
P. eq. H2O2 = (PM H2O2 : 2) = 17,008
Vol. eq. di ossigeno libero = (22.414 : 4) = 5603,5 ml
I percarbonati alcalini (esempio: K2C2O6 percarbonato di potassio) vengono determinati aggiungendone un campione esattamente pesato circa g 0,2-0,3) ad una soluzione formata da circa 250 ml di acqua distillata e 10 ml di H2SO4 conc. fredda: si sviluppa acqua ossigenata titolabile immediatamente come sopra:
K2C2O6 + H2SO4 þ K2SO4 + 2 CO2 + H2O2
P. eq. K2C2O6 = (PM K2C2O6 : 2) = 99,106 Volume equival. di
P. eq. Na2C2O6 = (PM Na2C2O6 : 2) = 83,007 ossigeno libero = ml 5603,5
I perborati alcalini (esempio: NaBO3 . 4 H2O perborato di sodio) vengono determinati aggiungendone un campione esattamente pesato (circa g 0,15-0,2) in una soluzione di circa 250 ml di acqua distillata e 10 ml di H2SO4 conc. fredda: si sviluppa acqua ossigenata titolabile immediatamente come sopra:
2 NaBO3 + H2SO4 + 4 H2O þ NaSO4 + 2 H3BO3 + 2 H2O2
P. eq. NaBO3 . 4 H2O = (PM NaBO3 . 4 H2O : 2) = 76,94
(Vol. equiv. di ossigeno libero = ml 5603,5)
DETERMINAZIONE DEL CROMO Cr METODO AL PERSOLFATO
Il persolfato ammonico (NH4)2S2O8, che ha numero di ossidazione 2 perché in soluzione libera un O [(NH4)2S2O8 + H2O þ (NH4)2SO4 + H2SO4 + O] è capace di ossidare il Cr3+ a Cr6+ in soluzione acida e cioè trasformare i sali cromici in acido cromico:
Cr2(SO4)3 + 3 ((NH4)2S2O8 + 8 H2O þ 2 H2CrO4 + 3 (NH4)2SO4 + 6 H2SO4
Il potenziale di ossidoriduzione normale di questa reazione è E°pers - E°Cr = 1,9 - 1,3 = 0,6 volt, cioè la reazione è abbastanza spinta.
Eliminato l'eccesso di persolfato per ebollizione (si svolge O2), si tratta l'acido cromico con un eccesso noto di soluzione di solfato ferroso; l'ac. cromico, che ha n. di ossidazione 3, ossida parte del Fe++ aggiunto:
6 FeSO4 + 2 H2CrO4 + 6 H2SO4 þ 3 Fe2(SO4)3 + Cr2(SO4)3 + 8 H2O
Titolando con KMnO4 N/10 f.c. noto l'eccesso di Fe++, si ottiene per differenza i ml corrispondenti al Cr in esame il cui equivalente (Cr6+/Cr3+) è Cr/3.
N.B.: Questa determinazione richiede l'assenza di ioni alogenidrici.
Reagenti necessari:
KMnO4 N/10 f.c. noto FeSO4 N/10 H2SO4 conc.
AgNO3 al 5% (NH4)2S2O8 al 20% H2SO4 al 10%
Modo di operare:
La soluzione in esame ovvero la parte aliquota su cui si opera, dovrà contenere il Cr su valori intorno a g 0,05. Si porta tale soluzione in un pallone da mezzo litro, si diluisce sui 220-250 ml con acqua dist., si aggiungono 50 ml di H2SO4 1:3 e 5 ml di AgNO3 al 5% che funziona da catalizzatore di ossidazione e da eliminatore di piccole quantità di ioni alogenidrici eventualmente presenti. Si aggiungono quindi 20 ml di persolfato ammonico (NH4)2S2O8 al 20%, si chiude il pallone con un imbutino e si fa bollire quietamente per 15-20 minuti.
Si prepara intanto una soluzione di FeSO4 circa N/10 (g 27,8 di FeSO4 -7 H2O oppure g 39,2 di sale di Mohr (NH4)2SO4 . FeSO4 . 6 H2O per litro) e leggermente acida per H2SO4; su 20 ml esatti di tale soluzione aggiunti di 10-20 ml di H2SO4 al 10% si opera una titolazione con KMnO4 N/10 f.c. noto notando la corrispondenza (1 ml soluzione Fe++ = n ml di KMnO4 N/10 f.c. noto).
Alla soluzione fredda contenente il Cr ossidato, si aggiungono 50 ml esatti di soluzione ferrosa e si titola l'eccesso ferroso col KMnO4 N/10 f.c. noto. Essendo che i 50 ml di soluz. Fe++ corrispondono a 50.n ml di KMnO4 di titolazione, la differenza 50.n - ml di . titolazione (non corretti) ci dà i ml corrispondenti al Cr che, corretti mediante moltiplicazione per il f.c. del KMnO4 N/10 usato, servono ad impostare la proporzione:
1.000 : P. eq./10 = ml risultati : X
P. eq. Cr = Cr/3 = 17,34 P. eq. = Cr2O3/6= 25,337
DETERMINAZIONE DELLO STAGNO Sn++
Una soluzione stannosa deve essere necessariamente notevolmente cloridrica; diversamente il Sn++ non rimane in soluzione ma si idrolizza e precipita. Tale soluzione acida di Sn++ può essere titolata direttamente a freddo con KMnO4 N/10 f.c. noto:
5 SnCl2 + 2 KMnO4 + 16 HCl þ 5 SnCl4 + 2 KCl + 2 MnCl2 + 8 H2O
Onde evitare di perdere il viraggio a causa dell'HCl presente che, una volta che si sia esaurito il Sn++ continuerebbe a ridurre il KMnO4. si aggiunge un grano di ioduro di potassio KI; essendo il potenziale di ossidoriduzione del KMnO4/I- inferiore a quello del KMnO4/Sn++, ma superiore a quello del KMnO4/Cl-, il KMnO4 dopo aver ossidato il Sn++, ossida il I- a I2 che colorerà in azzurro un ml di salda d'amido previamente aggiunto. (vedi pag. 38)
Reagenti necessari:
KMnO4 N/10 f.c. noto KI solido salda d'amido
Modo di operare:
La soluzione in esame ovvero l'aliquota in esame, dovrà contenere un intorno di g 0,18 di Sn++.
Alla soluzione fredda, notevolmente acida per HCl, si aggiunge un grano di ioduro di potassio KI ed un ml di salda d'amido Si titola con KMnO4 N/10 f.c. noto fino a viraggio viola.
1.000 : P. eq./10 = ml usati e corretti : X X = g di Sn
P. eq. Sn/2 = Sn = 59,35 P. eq. SnO2 = SnO2/2 = 75,35
GENERALITA' - PREP. SALDA D'AMIDO - PREP. SOLUZ. TIOSOLFATO SODICO N/10 E DET. FATT. CORR. (SOST. BASE: PERMANGANATO DI POTASSIO OPPURE BICROMATO DI POTASSIO) - DET. CLORO ATTIVO - DET. DEL Cu - DET. DELL'ACQUA OSSIGENATA - DET. DEL GLUCOSIO (FEHLING).
La iodometria è un metodo di analisi volumetrica con il quale si possono determinare tutte le quelle sostanze capaci di ossidare lo ioduro di potassio a Iodio (2 I- ----> I2 E° = + 0,53), cioè quelle sostanze che lo seguono nella serie elettrochimica. Lo Iodio che si libera in quantità equivalente alla sostanza ossidante in esame, viene poi titolato col tiosolfato sodico Na2S2O3 . 5 H2O che fra le varie sostanze capaci di ridurre lo Iodio è la più utile:
2 Na2S2O3 + I2 þ 2 NaI + Na2S4O6

Come si vede, il tiosolfato è un riducente monovalente ed il suo peso equivalente coincide con quello molecolare.
Per segnalare la scomparsa dello I2 si usa, come indicatore, la salda d'amido infatti da con lo Iodio un composto di adsorbimento azzurro scuro e tale colorazione scompare quando lo I2 sarà stato completamente ridotto ad I-.
PREPARAZIONE DELLA SALDA D'AMIDO
La salda d'amido è la soluzione acquosa dell'amido (parte solubile) ed ha la proprietà di adsorbire iodio e colorarsi in azzurro: con l'eliminazione dello iodio, essa si decolora. Conviene usarla sempre verso la fine delle reazioni iodometriche per evitare che assorba troppo iodio, che poi cede piuttosto lentamente.
Se, con lo iodio, si colora in verde o viola, significa che e alterata per fermentazione e va sostituita.
Modo di operare:
Un g circa di amido solubile viene spappolato in un mortaio con poca acqua fredda è versato lentamente ed agitando in 100 ml di acqua bollente, si aggiungono 2-3 g di KI e si conserva in bottiglia precedentemente bollita (salda l'amido).
PREPARAZIONE DELLA SOLUZIONE DI TIOSOLFATO SODICO N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO (sostanza base permanganato di potassio N/10 oppure bicromato di potassio)
Modo di operare:
Il peso equivalente del tiosolfato sodico Na2S2O3 . 5 H2O eguale al PM) è 248,20; la soluzione N/10 ne dovrà perciò contenere g 24,82 per litro.
Si pesano pertanto g 25 di tiosolfato sodico, si portano in pallone tarato da 1000 ml e si porta a volume con acqua distillata.
Si lascia a se la soluzione in bottiglia (o ambiente) scura per qualche giorno, affinchè la soluzione si stabilizzi di fronte all'acqua.
La soluzione così preparata e stabilizzata, deve essere controllata mediante una prova in bianco su una quantità esattamente nota di iodio che viene prodotta trattando una soluzione acida di KI con quantità esattamente note di ossidanti (permanganato KMnO4, bicromato K2Cr2O7, ecc.).
Modo di operare
:Sostanza base permanganato di potassio N/10
In un beaker si pongono circa 200 ml di acqua distillata fredda, 3-4 g di ioduro di potassio KI, 10 ml di acido solforico al 10 % e, a freddo, 20 o 25 ml esatti di KMnO4 N/10 f.c. noto: si libera una equivalente quantità di iodio:
2 KMnO4 + 10 KI + 8 H2SO4 þ 6 K2SO4 + 2 MnSO4 + 8 H2O + 5 I2
Si titola tale soluzione col tiosolfato sodico in esame; i ml teorici sono quelli di KMnO4 N/10 moltiplicati per il relativo f.c.; i ml pratici risultano dalla titolazione, aggiungendo la salda d’amido verso la fine della titolazione quando la soluzione tende a perdere il colore giallo dello iodio.
ml teorici / ml pratici = fattore correttivo
La soluzione di tiosolfato, avrà perciò la seguente denominazione:
Na2S2O3 N/10 f.c. . .
Ogni qualvolta sarà usata questa soluzione, bisognerà moltiplicarne i ml usati per il f.c.
Sostanza base bicromato di potassio
g 0,75 circa, pesati esattamente, di bicromato di potassio puro, vengono portati al volume di 250 ml esatti in matraccio tarato con acqua distillata.
In un beaker si pongono circa 200 ml di acqua distillata fredda, 3-4 g di ioduro di potassio KI, 10 ml di acido solforico al 10% e, sempre a freddo, 50 ml esatti di soluzione di bicromato preparata; si agita ed il bicromato libera una equivalente quantità di iodio:
K2Cr2O7 + 6 KI + 7 H2SO4 þ 4 K2SO4 + Cr2(SO4)3+ 7 H2O + 3 I2
Il bicromato libera 6 equivalenti di iodio e perciò il suo peso equivalente è
K2Cr2O7 /6 = 49,035
Si titola con il tiosolfato sodico in esame e verso la fine della reazione (la soluzione tende a perdere il colore giallo dello iodio) si aggiunge un ml di salda di amido (colorazione azzurra) e si completa la titolazione fino a decolorazione.
I ml usati in tale operazione sono i ml pratici.
Invece, dai g di bicromato presenti nei 50 ml prelevati (Q) si calcolano i ml teorici.
1.000 : 49,035/10 = X : Q X = ml teorici
ml teorici /ml pratici = fattore correttivo
La soluzione di tiosolfato preparata, avrà perciò la seguente denominazione:
Na2S2O3 N/10 f.c. . . . . . . .
Ogni qualvolta sarà usata questa soluzione, bisognerà moltiplicarne i ml usati per il f.c.
DETERMINAZIONE DEL CLORO ATTIVO (ACQUA DI CLORO E IPOCLORITI)
L'acqua di cloro (Cl2 + H2O þ HCl + HClO), ed anche la analoga acqua di bromo, sono capaci di liberare iodio dal KI secondo l'equazione:
C12 + 2 KI þ 2 KCI + I2
e lo iodio liberato (equivalente al cloro) viene titolato con tiosolfato sodico Na2S2O3 N/10 f.c. noto alla salda d'amido.
Gli ipocloriti, sono sostanze capaci di sviluppare cloro sotto l'azione di molti acidi anche deboli come il carbonico. In analitica, si usa HCl :
Il cloro che si sviluppa, viene detto cloro attivo e la sua determinazione ha grande importanza industriale. Questo cloro attivo è capace di mettere in libertà una equivalente quantità di iodio dal KI, e lo iodio verrà titolato con Na2S2O3 N/10 f.c. noto alla salda d'amido.
Reagenti necessari:
Na2S2O3 N/10 f.c. noto KI solido HCl al 10% salda d'amido
Modo di operare:
Determinazione del Cl2 sull'acqua di cloro:
In un flacone da 200-250 ml munito di tappo smeriglio, si introducono circa 100 ml di acqua distillata e 10 ml di HCI al 10% ottenendone la soluzione fredda.
Si introducono con una pipetta 20 ml di acqua di cloro, si chiude il tappo e si agita a lungo.
Si titola infine, dentro il flacone, con Na2S2O3 N/10 f.c. noto fino a che il colore giallo e quasi scomparso: a questo punto si aggiunge un ml di salda d'amido e si prosegue la titolazione fino a decolorazione completa.
P. eq. Cl2 = (PM Cl2: 2) = 35,457
Vol. eq. Cl2 = (22.414: 2) = ml 11.207
P. eq. Br2 = (PM Br2 : 2) = 79,916
Determinazione del cloro attivo sul cloruro di calce:
5 g di cloruro di calce, pesati in un pesafiltri, vengono portati in un matraccio tarato da 500 ml e portati a volume con acqua distillata.
Si sbatte a lungo la sospensione e quindi se ne prelevano rapidamente 50 ml calandoli dentro una soluzione fredda formata da 3 g circa di KI in 100 ml di acqua distillata e 10 ml HCl 10%.
Si agita a lungo e si titola con Na2S2O3 N/10 10 f.c. noto fino a quasi scomparsa del colore giallo: si aggiunge un ml di salda d'amido e si conclude la titolazione con la decolorazione della stessa
P. eq. Cl2 = (PM Cl2 : 2) = 35,457
Vol. eq. Cl2 = (22.414 : 2) = ml 11.207
DETERMINAZIONE DEL RAME
I sali rameici, reagiscono col KI formando il sale rameoso insolubile Cu2I2: a questa riduzione del rame, corrisponde formazione di iodio:
2 CuSO4 + 4 KI þ Cu2I2 + 2 K2SO4 + I2
La reazione decorre completamente verso destra operando in un eccesso di KI ed in ambiente mediamente solforico.
L'eccesso di KI (piuttosto costoso) può essere sostituito con la quantità stechiometrica o poco più di KI ed un eccesso di solfocianuro di potassio KCNS anch'esso capace di precipitare il rame come sale rameoso Cu2(CNS)2 ancora meno solubile dello ioduro Cu2I2.
2 CuSO4 + 2 KI + 2 KCNS þ Cu2(CNS)2 + 2 K2SO4 + I2
In ogni caso, lo iodio liberato viene titolato con Na2S2O3 N/10 f.c. noto.
Reagenti necessari:
H2SO4 al 10% salda d'amido Na2S2O3 N/ 10 f.c. noto KI solido (KCNS, solido)
Modo di operare:
La soluzione, contenente approssimativamente una quantità di rame Cu++ corrispondente a 30-40 ml di soluzione N/10 (g 0,20 - 0,25 di Cu), viene posta in un beaker da 400 ml in volume di 200-250 ml. Si aggiungano 10 ml di H2SO4 al 10% e 4 g di KI previamente sciolti in 50-100 ml di acqua distillata (oppure 1 g di KI e 3 di KCNS unitamente sciolti in acqua distillata) e si agita a lungo.
Si titola con Na2S2O3 N/10 f.c. noto; verso la fine si aggiunge un ml di salda d'amido e si conclude la titolazione fino a decolorazione della stessa persistente almeno 5': rimarrà il colore del precipitato di Cu2I2 biancastro (oppure del Cu2(CNS)2 grigio rosato se si usa KCNS).
P. eq. Cu (PA Cu) = 63,57
P. eq. CuSO4 . 5 H2O = (PM CuSO4 . 5 H2O) = 249,71
DETERMINAZIONE DELL'ACQUA OSSIGENATA
L'acqua ossigenata, e capace di liberare iodio dal KI secondo l'equazione:
H2O2 + 2KI + H2SO4 þ K2SO4 + 2 H2O + I2
La reazione decorre bene verso destra in ambiente discretamente acido ed aggiungendo qualche goccia di molibdato di ammonio in soluzione al 5%, che agisce da catalizzatore, essendo la reazione piuttosto lenta.
Reagenti necessari:
Na2S2O3 N/ 10 f.c. noto KI solido salda d'amido H2SO4 al 20%
(NH4)2MoO4 al 7%
Modo di operare:
L'acqua ossigenata in esame, viene diluita con misurate quantità onde averla sullo 0,5% ossia sui 1,6 volumi.
Si prepara una soluzione dentro un flacone munita di tappo smeriglio, contenente 100 ml di acqua distillata, 1-2 g di KI e 20 ml di H2SO4 10%. Dentro tale soluzione fredda, si aggiungono 3 gocce molibdato ammonico (NH4 )2MoO4 al 7% ed infine , si calano 10 ml esatti dell'acqua ossigenata diluita come sopra. Si chiude il tappo e si lascia a se per 10’.
Si titola quindi con Na2S2O3 N/10 f.c. noto chiudendo frequentemente la bottiglia fino a quasi decolorazione, poi si aggiunge un ml di salda d'amido e si conclude la titolazione fino a decolorazione della stessa. Operare lentamente.
P. eq. H2O2 = (PM H2O2 :2) = 17,008
Vol. eq. H2O2 = (22.414 : 4) = ml 5603,5 di ossigeno libero
DETERMINAZIONE DEL GLUCOSIO FEHLING
Questa determinazione si basa sull'azione riduttiva del glucosio sul rame Cu++ che precipita come Cu2O2 sulla determinazione quantitativa dell'ossido rameoso precipitato; dalla quantità di rame trovata si deduce la quantità di glucosio in esame, non per via stechiometrica, perché l'azione riduttiva del glucosio non e stechiometrica, ma consultando la tabella sperimentale riportata in seguito; la tabella e valida soltanto se si opera nelle condizioni indicate o comunque scostandosi da esse assai poco.
Il reattivo Fehling A contiene il CuSO4 destinato ad essere ridotto dal glucosio; siccome tale riduzione si verifica bene in ambiente basico, il Fehling B contiene appunto NaOH nonché il sale di Seignette che complessando il Cu++ impedisce la precipitazione di CuO per effetto della NaOH mentre precipiterà soltanto il Cu2O originato dalla riduzione del glucosío sul Cu++.
Reagenti necessari:
CuSO4 . 5 H2O Sale di Seignette NaOH solido HNO3 conc. NH3 al 10% H2SO4 al 10% KI solido (KCNS) Na2S2O3 N/10 f.c. noto salda d'amido
Modo di operare:
Preparazione del liquido di Fehling: è composto di due distinte soluzioni:
Fehling A: g 69,278 di CuSO4. 5 H2O sciolto in acqua e portato al litro.
Fehling B: g 346 di sale di Seignette (tartrato sodico potassico) sciolti in acqua + 100 g di idrato sodico solido in gocce e portare il tutto al litro.
Le due soluzioni vanno conservate in bottiglie separate e, al momento dell'uso vengono prelevate a volumi uguali.
Determinazione del glucosio
La soluzione contenente il glucosio in esame deve essere portata a volume esatto in modo da contenere non più del 1 %.
In un beaker si pongono 30 ml esatti di Fehling A e 30 ml esatti di Fehling B nonchè 60 ml di acqua distillata; si porta all'ebollizione e vi si aggiungono 25 ml esatti di soluzione di glucosio e si fa bollire per 2’ (controllarli); si toglie la fiamma, si aggiungono 100 ml di acqua fredda, si filtra l'ossido rameoso rosso formatosi su fascia bianca e si lava a lungo con acqua calda.
Si pone sotto l'imbuto un beaker pulito, si fora il filtro con la bacchetta e si fa cadere il precipitato nel beaker con acqua calda; si lava bene il filtro con 20 ml circa di HNO3 dil. ed ancora con acqua. Si aggiunge nel beaker 2-3 ml di HNO3 conc. e si scalda i il sotto cappa a debole fiamma: si sviluppano vapori nitrosi perché il Cu2O viene ossidato a Cu(NO3)2 dall'ac. nitrico:
3 CuO + 2 HNO3 þ 6 CuO + H2O + 2 NO
6 CuO + 12 HNO3 þ 6 Cu(NO3)2 + 6 H2O
3Cu2O + 14 HNO3 þ 6 Cu(NO3)2 + 7 H2O + 2 NO
Ottenuta la soluzione, si aggiunge ammoniaca diluita fino a colore azzurro e quindi H2SO4 al 10% goccia a goccia fino a schiarire la soluzione e poi ancora 10 ml; si aggiungono allora 4 g di KI oppure 1 g di KI e 3 g di KCNS, sciolti previamente in poca acqua distillata e si determina il Cu iodometricamente (vedi pag. 41).
Dai mg di Cu trovati, si calcola il glucosio presente nei 25 ml esaminati, consultando la seguente tabella (Allihn).
|
Cu |
GLUCOSIO |
Cu |
GLUCOSIO |
Cu |
GLUCOSIO |
Cu |
GLUCOSIO |
|
10 |
6,1 |
125 |
63,7 |
240 |
123,9 |
355 |
187,2 |
|
15 |
8,6 |
130 |
66,2 |
245 |
126,6 |
360 |
190,0 |
|
20 |
11,0 |
135 |
69,8 |
250 |
129,2 |
365 |
192,9 |
|
25 |
13,5 |
140 |
71,3 |
255 |
131,9 |
370 |
195,7 |
|
30 |
16,0 |
145 |
73,9 |
260 |
134,6 |
375 |
198,6 |
|
35 |
18,5 |
150 |
76,5 |
265 |
137,3 |
380 |
201,4 |
|
40 |
20,9 |
155 |
79,1 |
270 |
140,0 |
385 |
204,3 |
|
45 |
23,4 |
160 |
81,7 |
275 |
142,8 |
390 |
207,1 |
|
50 |
25,9 |
165 |
84,3 |
280 |
145,5 |
395 |
210,0 |
|
55 |
28,4 |
170 |
86,9 |
285 |
148,3 |
400 |
212,9 |
|
60 |
30,8 |
175 |
98,5 |
290 |
151,0 |
405 |
215,8 |
|
65 |
33,3 |
180 |
92,1 |
295 |
153,8 |
410 |
218,7 |
|
70 |
35,8 |
185 |
94,7 |
300 |
156,5 |
415 |
221,6 |
|
75 |
38,3 |
190 |
97,3 |
305 |
159,3 |
420 |
224,5 |
|
80 |
40,8 |
195 |
100,0 |
310 |
162,0 |
425 |
227,5 |
|
85 |
43,4 |
200 |
102,6 |
315 |
164,8 |
430 |
230,4 |
|
90 |
45,9 |
205 |
105,3 |
320 |
167,5 |
435 |
233,4 |
|
95 |
48,4 |
210 |
107,9 |
325 |
170,3 |
440 |
236,3 |
|
100 |
50,9 |
215 |
110,6 |
330 |
173,1 |
445 |
239,3 |
|
105 |
53,5 |
220 |
113,2 |
335 |
175,9 |
450 |
242,2 |
|
110 |
56,0 |
225 |
115,9 |
340 |
178,7 |
455 |
245,2 |
|
115 |
58,6 |
230 |
118,5 |
345 |
181,5 |
460 |
248,1 |
|
120 |
61,1 |
235 |
121,2 |
350 |
184,3 |
465 |
251,1 |
GENERALITA' - PREP. SOLUZ. BROMATO DI POTASSIO N/10 E DET. FATTORE CORR. -DET. DEL FENOLO (E ANILINA) DET. DEL Al CON 8-OSSICHINOLINA DET. DEL Sb (As e Sn
).
Il bromato di potassio KBrO3 e un ossidante in soluzione acida con numero di ossidazione 6:
5+ 1-
KBrO3 þ KBr + 3 O (BrO3-)/Br- E° = 1,48 V
ma, ciò che interessa particolarmente, è una sostanza bromurante quando, sempre in soluzione acida, venga a reagire col bromuro di potassio KBr.
La bromurazione è un'operazione che nei riguardi delle sostanze aromatiche, e di facile esecuzione e di andamento quantitativo; queste bromurazioni vengono condotte in soluzione acida usando bromuro e bromato di potassio, che sviluppano bromo:
5 KBr + KBrO3 + 3 H2SO4 þ 3 K2SO4 + 3 H2O + 3 Br2
Usando su quantità in eccesso il bromuro KBr, quantità esatte di bromato KBrO3 si ottengono quantità equivalenti e note di bromo Br2: il bromo prodotto sarà parzialmente consumato nella bromurazione di un determinato composto, mentre quello in eccesso, sarà convertito in iodio mediante aggiunta di ioduro di potassio.
Br2 + 2 KI þ 2 KBr + I2
e lo iodio, sarà titolato con tiosolfato sodico Na2S2O3 N/10 f.c. noto.
PREPARAZIONE DELLA SOLUZIONE DI KBrO3 N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO
Dalla reazione di formazione del bromo, da bromuro e bromato, risulta che il peso equivalente del bromato di potassio è
KBrO3 /6 = 167,01/6 = 27,8353
Modo di operare:
Per preparare un litro di soluzione N/10 si pesa esattamente la decima parte dell'equivalente e cioè g 2,7835 di KBrO3 e si sciolgono in acqua distillata in matraccio tarato da litro portandoli a volume esatto.
Essendo il bromato una sostanza stabile, usando un prodotto puro non è necessario determinare il fattore correttivo, ma volendolo determinare, si pone in una bottiglia da 1/2 litro 200 ml di acqua distillata, circa 2 g di KBrO3 20 ml esatti di KBrO3 N/10 10 in esame, 10 ml di H2SO4 10% a freddo, si chiude il tappo e si agita per qualche minuto ottenendo lo sviluppo di bromo. Si aggiungono quindi 3 g circa di KI previamente sciolti in poca acqua lavando il tappo con quest'ultima soluzione; si chiude il tappo e si agita ottenendo la conversione del bromo in iodio. Quindi si titola con tiosolfato sodico Na2S2O3 N/10 f.c. noto alla salda d'amido:
fatt. corr. = ml teorici / ml pratici 20 ml usati in titolazione
DETERMINAZIONE DEL FENOLO (E ANILINA)
Il bromo, agisce sul fenolo nel seguente modo:

ed il peso equivalente del fenolo sarà 94,109/6 = C6H5OH /6= 15,685.
Mentre il bromofenolo precipita, l'eccesso di bromo viene trasformato in iodio mediante KI e, lo iodio corrispondente, titolato col Na2S2O4 N/ 10 f.c. noto.
N.B. La quantità di fenolo da determinare dovrà essere utilmente mantenuta sui 0,04 g.
Reagenti necessari:
KBrO3 N/10 f.c. noto KBr solido HCl conc. KI solidoNa2S2O3 N/10 f.c. noto salda d'amido
Modo di operare:
La soluzione in esame contenente fenolo viene, se necessario, portata a volume esatto allo scopo di operare sulla parte aliquota che contenga attorno a g 0,04 di fenolo.
Tale aliquota viene portata in una bottiglia a tappo smeriglio; si aggiungono 2 g circa di KBr e 50 ml esatti di KBrO3 N/10 aggiungere 5 ml di HCl conc., tappare la bottiglia, sbattere per 1’ e lasciare a se per 20' ottenendo la precipitazione del tribromofenolo.
Nella bottiglia, eventualmente raffreddata sotto il filo dell'acqua corrente, si introducono g 2-3 di KI previamente sciolti in acqua, approfittando di tale introduzione per lavare il tappo. Si chiude la bottiglia, si sbatte per 1’ e si lascia a se per 10'. Si toglie il tappo, lo si lava sopra la bottiglia e si titola in bottiglia con Na2S2O3 N/10 f.c. noto; verso la fine della titolazione si aggiunge un ml di salda di amido e si prosegue fino a viraggio.
(50 - ml usati in titolazione) = ml N/10 corrispondenti al fenolo
1.000 : 1,5685 = ml risultati: X X g di fenolo presenti nell'aliquota esaminata
L'anilina C6H5NH2 può essere determinata esattamente allo stesso modo operando su un campione che contenga circa g 0,04 di anilina.
Il peso equivalente è 93,125/6 = 15,521
DETERMINAZIONE DELL'ALLUMINIO CON 8-OSSICHINOLINA
La 8-ossichinolina è un fenolo capace di precipitare l'Al+++ (ed anche il Mg, Cu, Zn, Mn, ed altri) come ossichinolato di alluminio Al(ox)3 giallo; questo, filtrato lavato e ridisciolto in acido forte, rimette in libertà l'ossichinolina stechiometrica:

L'ossichinolina, viene bromurata col bromo prodotto dal bromuro e bromato di potassio:

Infine, si converte in iodio il bromo di eccesso e si titola lo iodio con tiosolfato sodico N/10 f.c. noto.
Reagenti necessari:
KBrO3 N/10 f.c. noto KBr solido 8-ossichinolina
CH3COOH al 25 % CH3COONH4 HCl al 10%
NH3 al 5% KI Na2S2O3 N/10 f.c. noto salda d'amido
Modo di operare:
Anzitutto si prepara una soluzione di 8-ossichinolina al 5% in acido acetico al 25%. La soluzione in esame, tutta oppure una parte aliquota nota che contenga non oltre g 0,018 di Al, viene diluita a 100 ml circa con acqua a calda; si aggiunge acetato ammonico in soluzione concentrata fino a intorbidamento di Al(OH)3 e quindi HCl al 10% goccia a goccia fino a scioglimento. Si ottiene così una soluzione appena acida nonchè tamponata di fronte all'acido forte che si libererà nella precipitazione seguente del ossichinolato di alluminio.
Si scalda a 60° C e si aggiungono 10-20 ml di reattivo 8-ossichinolina; si completa (o si provoca) la precipitazione aggiungendo goccia a goccia NH3 al 5 % (pH 4 circa). Ottenuto il precipitato si lascia a se a freddo per 20'; quindi si filtra su fascia bianca e si lava bene con acqua calda per asportare l'ossichinolina in eccesso (usare circa 300 ml di acqua calda a piccole aliquote).
Porre sotto l'imbuto un beaker pulito, forare il filtro con una bacchetta e portare giù gran parte del precipitato con circa 50 ml di acqua bollente; quindi lavare bene il filtro con circa 50 ml di HCl 10% caldo e, se necessario, ancora con acqua calda (il filtro deve rimanere bianco); scaldare il liquido raccolto nel beaker ed agitare fino ad ottenere la perfetta soluzione (eventualmente aggiungere poco a poco HCl 10%). Passare tutta la soluzione in una bottiglia a tappo smeriglio lavando bene il bicchiere; si aggiungono a freddo 2 g di KBr e 100 ml esatti di KBrO3 N/ 10; si chiude la bottiglia e si sbatte per 2’: se non si forma precipitato di bromo-ossichinolina, lo si provoca aggiungendo acetato ammonico sciolto in pochissima acqua; ottenuto il precipitato, si lascia a se per 10'. Si aggiungono circa 2 g di KI sciolti in poca acqua (lavare il tappo introducendo tale soluzione), si sbatte a freddo e si lascia a se 10'. Infine si titola lo iodio liberatosi col Na2S2O3 N/10 f.c. noto aggiungendo un ml di salda d'amido verso la fine della titolazione.
(100 - ml usati in titolazione) = ml N/10 corrispondenti all'Alluminio
Essendo che un atomo di Al precipita 3 mol. di ossichinolina, e ciascuna mol. di ossichinolina pretende 4 Br nella bromurazione, il peso equivalente sarà Al/12 = 2,2475.
1.000 : 0,22475 = ml risultanti X = g di alluminio presenti nell'aliquota esaminata
DETERMINAZIONE DELL'ANTIMONIO (ARSENICO E STAGNO)
Una soluzione notevolmente acida per HCl contenente Sb3+ (o As3+ o Sn2+), può essere titolata direttamente a caldo con KBrO3 N/10 f.c. noto secondo la reazione:
3 SbCl3 + KBrO3 + 6 HCl þ 3 SbCl5 + KBr + 3 H2O
Dopo aver ossidato tutto l'antimonio da Sb3+ a Sb5+ , l'ulteriore goccia di KBrO3 agisce sul KBr formatosi nella reazione stessa con formazione di bromo libero Br,: quest'ultimo va a bromurare un indicatore previamente introdotto (rosso di metile, blu di timolo o altri) provocando un viraggio molto evidente.
Il rosso di metile e un acido debole e precisamente l'acido p-dimetilammino -azobenzen-ortocarbonico; esso in soluzione acida è rosso, colore dovuto alle sue molecole non dissociate; in am.. biente basico diventa giallo, colore dovuto alle sue molecole dissociate; la bromurazione nei punti indicati dalle X, esalta la forza dell'acido-indicatore in modo sufficiente da permettergli una certa dissociazione malgrado l'ambiente sia nettamente acido per HCl: la conseguenza è che il colore vira al giallo che è appunto il colore delle molecole dissociate. Il rosso di metile funziona bene nelle titolazioni del Sb3+ e As3+ ma non funziona con lo Sn2+ che lo riduce nel doppio legame azoico decolorandolo subito; per lo stagno si può utilmente usare il blu di timolo con viraggio rosso violaceo/rosa chiaro.
Reagenti necessari:
KBrO3 N/10 f.c. noto Rosso di metile HCl conc.
Modo di operare:
La soluzione in esame ovvero la parte aliquota su cui si opera, dovrà contenere intorno a g 0,18 di Sb3+ (g 0, 11 di As3+ e g 0, 17 di Sn2+).
La soluzione eventualmente aggiunta di HCl 1:2 viene scaldata sui 70°- 80° C°; si aggiungono due gocce di rosso di metile (soluz. alcolica 0,2%) rendendola nettamente rossa e si titola a caldo con KBrO3 N/10 10 f.c. noto fino a viraggio giallo incipiente.
N.B.
Nella determinazione dello stagno usare come indicatore il blu di timolo.1.000: P. eq./10 = ml usati e corretti : X
P. eq. Sb = Sb /2 = 60,88 P. eq. As = As /2 = 37,455
P eq. Sn = Sn /2 = 59,35
GENERALITA' - PREP. SOLUZ. EDTA N/io E DET. FATT. CORR. (SOST. BASE: SOLFATO DI ZINCO - DET. Al, Ba, Ca (IN PRES. DI Mg), Ca (Mg, Zn), SOLFATI DET. DUREZZA DELLE ACQUE - DET. DEL Cd (Zn) SU BAGNO GALVANICO
.L'analisi complessometrica si basa sull'uso di certe sostanze titolanti capaci di formare complessi con li ioni che si intendono determinare. Le più comunemente oggi usate tra queste sostanze complessanti sono l’acido nitriloacetico HN(CH2COO)2, l'acido etilendiamminotetracetico [CH2N(CH2COOH)2]2 EDTA ed il sale bisodico.
Questo acido dà ioni complessi con vari ioni (Ca, Mg, Zn, Mn, Cu, ecc.) del tipo chelato. I chelati sono complessi in cui un atomo metallico è legato a due punti diversi di una stessa molecola di reattivo, i due legami che formano la chela sono, uno di tipo elettrovalente (ionico) in cui il metallo sostituisce un idrogeno acido, l'altro è di tipo covalente dativo nel quale il doppietto elettronico che costituisce il legame covalente, appartiene ad uno solo degli atomi contraenti il legame:

Si vede che questo complesso col Ca presenta due legami chelati: ciascuna chela e formata da un legame ionico (linea intera: sostituzione del H) e da uno dativo (linea frecciata: il doppietto elettronico appartiene all'N).
Il meccanismo delle titolazioni complessometriche consiste nell'uso di adatti indicatori che aggiunti in piccola quantità alla soluzione contenente ione da determinare formano con questo una piccola quantità di complesso vistosamente colorato e di colore diverso dall'indicatore libero; titolando una tale soluzione, per es., con EDTA, questo complessa lo ione da determinare e, verso la fine della titolazione, anche quella piccola aliquota di ione che era legata all'indicatore; l'indicatore viene così liberato rivelando il suo colore che, come abbiamo detto, e diverso dal colore del suo complesso con lo ione:
Es.: Ca-ind + EDTA þ Ca-EDTA + ind.
rosso azzurro
Varie sostanze sono usate come indicatori utili per la determinazione dei diversi ioni e qui sotto sono elencati alcuni con indicate le modalità di uso, il loro colore ed il colore dei loro complessi con gli ioni che sono in grado di determinare.

Per realizzare i valori di pH previsti per le varie determinazioni, si usano soda caustica, acido acetico, ammoniaca oppure soluzioni tampone appositamente preparate; due molto utili sono qui sotto indicate:
Soluzione tampone pH 10 Soluzione tampone pH 4,6
g 60 di NH4Cl (1 mole circa) + CH3COOH 2 m + CH3COONa 2m
e ml 400 di NH3 conc. (6 moli 1:1 circa), il tutto portato al litro.
PREPARAZIONE DELLA SOLUZIONE DI EDTA N/10 E DETERMINAZIONE DEL FATTORE CORRETTIVO (sostanza base solfato di zinco eptaidrato)
Il peso molecolare del sale sodico del EDTA è 372,24, trattandosi di sostanza bivalente, una sua soluzione N/10 dovrà contenere g 18,612 di sale nel litro. Il sale è perfettamente solubile e, se era ben conservato non richiede nemmeno la determinazione del fattore correttivo.
Comunque questa determinazione si esegue preparando una soluzione a contenuto esattamente noto, intorno a g 14-15 di ZnSO4 . 7H2O (p. mol. 287,55) per litro, cioè circa N/10.
20 ml esatti di tale soluzione diluiti a circa 100 ml ed aggiunti di 10 ml circa di tampone pH 10 ed una punta di spatola di indicatore eriocromo-sale (vedi pag. 50); si titola con l'EDTA N/10 in esame fino a viraggio dal rosso a1 verde.
Reattivi necessari:
EDTA N/10 f.c. noto ZnSO4 N/10 f.c. noto Tampone pH 4,6
Indicatore ditizone Alcol Metilico
Modo di operare:
La soluzione in esame contenente non più di g 0,05 di Al viene aggiunta di 50 ml esatti di EDTA N/10 f.c. noto e portata sotto ebollizione per 10'.
Dopo raffreddamento si aggiunge circa altrettanto volume di alcol metilico e 10 ml di soluzione tampone pH 4,6 nonché 3-4 ml di indicatore ditizone (vedi pag. 50)
Si titola l’eccesso di EDTA con ZnSO4 N/10 f.c. noto fino a viraggio da bruno-violetto al rosa.
P. eq = Al/2 = 13,485
Reattivi necessari:
EDTA N/10 f.c. noto Alcol metilico NH3 conc.
Indicatore Porpora di ftaleina
Modo di operare:
La soluzione che deve contenere non più di g 0, 1 di Ba. viene portata sui 100 ml con acqua distillata ed aggiunta di circa 100 ml di alcol metilico 10 ml di ammoniaca conc. e 0,5 ml di indicatore porpora di ftaleina (vedi pag. 50).
Si titola con EDTA N/10 f.c. noto fino a viraggio dal rosso-violetto al violetto stinto.
DETERMINAZIONE DEL SOLO CALCIO (in presenza di Magnesio)
Reattivi necessari:
EDTA N/10 f.c. noto KOH al 25 %
Indicatore acido calcocarbonico
Modo di operare:
La soluzione deve contenere non più di g 0,025 di Ca e quantità non eccessive di Mg, vi si aggiungono 10 ml di KOH al 25% ottenendo la precipitazione del Mg come idrato ed un pH 12 circa, si aggiunge una punta di spatola di indicatore acido calconcarbonico (v. pag. 132) e si titola agitando energicamente con EDTA N/10 fino a viraggio dal rosso vivo all'azzurro.
P. eq. = Ca/2 = 20,04
DETERMINAZIONE DEL CALCIO (Magnesio, Zinco)
Reattivi necessari:
EDTA N/10 f.c. noto Tampone pH 10 Indicatore eriocromo – sale
Modo di operare:
La soluzione deve contenere non più di g 0,025 di Ca (oppure non più di g 0,015 di Mg e g 0,04 di Zn) viene diluita sui 100 ml e aggiunta di 10 ml di soluzione tampone pH 10. Si titola fino a viraggio dal rosso al verde con colorazione grigia intermedia.
N.B. Se sono presenti sia Ca che Mg che Zn essi vengono titolati insieme.
P. eq. = Ca/2 = 20,04
DETERMINAZIONE DEI SOLFATI
I solfati possono essere determinati volumetricamente precipitandoli con un eccesso noto di BaC12 e titolando il Ba++ eccedente con EDTA N/10 all'eriocromo.
Reattivi necessari:
EDTA N/10 f.c. noto HCl conc. BaCl2 . H2O N/10
Alcol metilico (o etilico) NH3 conc. Indicatore porpora di ftaleina
Modo di operare:
La soluzione in esame, ovvero l'aliquota su cui si opera, dovrà utilmente contenere intorno a g 0,1 di SO4--.
Si prepara una soluzione di BaC12 approssimativamente N/10 (BaC12 . 2H2O g 12,16 per litro); a 20 ml esatti di questa soluzione si aggiungono 20 ml circa di acqua dist., 40 ml di alcol metilico (o etilico), 5 ml di NH3 conc. e 2-3 gocce di indicatore porpora di ftaleina. Si titola con EDTA N/10 fino a viraggio dal rosso-violetto al violetto stinto; si nota la corrispondenza Ba++/EDTA (i ml di soluz. Ba++ = n. ml EDTA N/10) e si conserva questa soluzione virata per il confronto visivo del viraggio nella successiva titolazione. Alla soluzione in esame acida per HCl si aggiungono all'ebollizione 50 ml esatti di soluz. BaCl2 precedentemente preparata (precipitano i solfati), si fa bollire per 15 minuti e, dopo raffreddamento si titola con EDTA N/10 fino a viraggio. 50 n. (n = corrispondenza Ba++/EDTA) meno i ml usati in quest'ultima titolazione rappresenta i ml corrispondenti al SO4--.
P. eq. SO4-- = SO4-- /2 = 49
DETERMINAZIONE DELLA DUREZZA DELLE ACQUE
Reagenti necessari:
EDTA N/50 g 3,7224/litro tampone pH 10 indicatore eriocromo
Modo di operare:
Durezza totale (tutti i sali di Ca e Mg): si procede come indicato per la determinazione del Ca e si opera su 100 ml esatti di acqua in esame.
Si usa EDTA N/50 perchè essendo il calcolo
50.000 : 50,045 = ml usati : X (P. eq. CaCO3 = 50,045)
X = (50,045/50.000) . ml usati = (0,001 . ml usati) = g di CaCO3 in 100 ml di acqua in esame
50.000 e cioè ml usati = g di CaCO3 in 100 litri di acqua in esame
Risulta quindi che i ml usati corrispondono esattamente ai gradi di durezza Francesi (°F) che sono appunto g di CaCO3 per 100 litri.
N.B. Mn e Zn vengono titolati come durezza, se è presente Cu, bisogna complessarlo con poco KCN perchè disturba il viraggio, in presenza di Fe bisogna aggiungere poco Na2S.
Durezza permanente (solfati e cloruri di Ca e Mg): si fanno bollire per 15' 100 ml esatti di acqua in esame si filtra su fascia bianca raccogliendo il filtrato in pallone tarato da 100 ml e si porta a volume con acqua distillata attraverso il filtro. Si opera su tali 100 ml come detto precedentemente.
Durezza temporanea (bicarbonati di Ca e Mg) = durezza totale - durezza permanente.
Oppure viene determinata come indicato in acidimetria.
DETERMINAZIONE DEL CADMIO (ZINCO) SU UN BAGNO GALVANICO
I bagni galvanici contengono generalmente il Cadmio complessato con lo ione cianidrico: si tratta di soluzioni alcaline di cadmiocianuro di sodio in equilibrio con i suoi ioni; il complesso e molto stabile (equilibrio fortemente spostato a sinistra)
Cd(CN)4-- Ö Cd++ + 4CN-
e l’EDTA non è capace di titolare per complessazione il Cd++ finchè questo è prigioniero nel complesso cianidrico; quindi la possibilità di titolare il Cd++ col EDTA è collegata alla possibilità di decompolessare il Cd++ dal suo complesso cianidrico e ciò è realizzabile con aldeide formica che con HCN forma una cianidrina:
Liberato in questo modo il Cd++, esso e titolabile col EDTA. Per trasformare l'ambiente nettamente alcalino del bagno galvanico con l'ambiente ammoniacale pH 10 necessario in complessometria bisogna neutralizzare il bagno con acido solforico fino a leggero intorbidamento dovuto al formarsi dello idrato di Cadmio e quindi operare col tampone pH 10 e l'eriocromo previsti dal metodo.
Reattivi necessari
:EDTA N/10 f.c. noto H2SO4 10% Tampone pH 10
Indicatore eriocromo-sale Formaldeide 40%
Modo di operare:
La soluzione in esame ovvero la parte aliquota su cui si opera dovrà utilmente contenere intorno a g 0,22 di Cd (g 0, 12 di Zn).
Alla soluzione in esame si aggiunge goccia a goccia H2SO4 al 10% fino a leggero intorbidamento, quindi 10 ml di tampone pH 10, una punta di spatola di eriocromo-sale e circa 5 ml di formaldeide al 35-40% (colorazione rosa). Si titola col EDTA N/10 fino a viraggio azzurro-verde e si accerta che il viraggio non retroceda aggiungendo qualche ml di formaldeide (se ciò si verifica, si continua la titolazione).
P. eq. Cd = Cd /2= 56,205 P. eq. Zn = /2 = 32,69
